- Rosetta@home
-
Rosetta@home
Rosetta@home Desarrollador Baker laboratory, University of Washington; Rosetta Commons http://boinc.bakerlab.org/rosetta Información general Lanzamiento inicial 6 de octubre de 2005 Última versión estable Rosetta Beta: 5.98, Rosetta Mini: 1.98
25 de junio de 2008Sistema operativo Multiplatforma Plataforma BOINC Licencia Libre para uso académico y sin ánimo de lucro, licencia propietaria disponible para uso comercial[1] Estado actual Activo Rosetta @ home es un proyecto de computación distribuida para la predicción de estructura de proteínas que se ejecuta sobre la plataforma BOINC. El proyecto es administrado por el laboratorio Baker de la Universidad de Washington. Apunta ser una plataforma de acople proteína-proteína y a diseñar nuevas proteínas con la ayuda de más de 86000 computadoras voluntarias procesando cerca de 77 teraFLOPS promedio, de acuerdo a lo medido el 30 de noviembre de 2008.[2] El videojuego Foldit de Rosetta@home apunta a lograr este objetivo con una aproximación crowdsourcing. Aunque gran parte del proyecto está orientado hacia la investigación básica sobre la mejora de la precisión y robustez de los métodos de la proteómica, Rosetta @ home también hace investigación aplicada sobre la malaria, el mal de Alzheimer y otras patologías.[3]
Al igual que todos los proyectos BOINC, Rosetta @ home utiliza recursos inactivos del procesador de las computadoras voluntarias para ejecutar cálculos en unidades de trabajo individuales. Los resultados finalizados son enviados a un servidor central del proyecto, donde se validan y son asimilados en bases de datos. El proyecto es multiplataforma, y se ejecuta en una amplia variedad de configuraciones de hardware. Los usuarios pueden ver el progreso de cada una de sus proteínas en la predicción de estructura del protector de pantalla Rosetta @ home. Además de la investigación relacionada con las enfermedades, la red Rosetta@home sirve como un marco de prueba para los nuevos métodos de bioinformática estructural. Estos nuevos métodos se utilizan en otras aplicaciones basadas en Rosetta, como RosettaDock y Human Proteome Folding Project, después de haber sido desarrollado y probado suficientemente su estabilidad en un gran número de computadoras de diferentes arquitectura que participan del proyecto.
Dos pruebas importantes para los nuevos métodos desarrollados en Rosetta@home son los experimentos Evaluación critica de las técnicas para la predicción de la estructura de la proteína (CASP) y la Evaluación critica de la predicción de la interacciones (CAPRI), se desarrollan cada 6 meses para evaluar el estado de la técnica en la predicción de la estructura de la proteína y la predicción de la estación de acople proteína-proteína.
Rosetta@home se encuentra siempre entre los principales predictores de acoplamiento y es uno de los mejores predictores de estructura terciaria disponible.[4]
El nombre de este proyecto se debe un artefacto egipcio antiguo denominado piedra de Rosetta.
Contenido
Plataforma informática
Hay versiones tanto de Rosetta@home como de la plataforma BOINC disponibles tanto para Linux, para Windows, para Macintosh y otras plataformas.[5] Los requerimientos mínimos para participar del proyecto una CPU con un mínimo de 500 KHz de velocidad, 200 megabytes de espacio libre en disco, 256 megabytes de memoria RAM y conexión a Internet.[6] A partir del 1 de diciembre de 2008 se utiliza la versión 5,98 de Rosetta,[7] y la 6.2.19 recomendada para BOINC.[5] La aplicación utiliza el puerto 80 de HTTP para comunicarse con los servidores de la Universidad de Washington, y durante el intercambio de contraseñas se utiliza el puerto HTTPS 443. Para el control remoto y local el cliente BOINC utiliza los puertos 31416 y 1043, los cuales debieran estar desbloqueados si existe un firewall en la red.[8]
Las unidades de trabajo (Workunits) contienen datos de proteínas individuales que están distribuidos en servidores ubicados desde el laboratorio Baker de la Universidad de Washington hasta en las computadoras voluntarias y lo que hacen es calcular una predicción de estructura para la proteína asignada. Para evitar predicciones de estructura duplicadas lo que se hace es generar un valor denominado semilla aleatoria.[9] Esto le da a cada predicción una trayectoria única de descenso a lo largo del plano energético de la proteína.[10] Las predicciones de estructura de la proteína de Rosetta@home son una aproximación de un valor mínimo en un plano energético de una proteína. Este mínimo global representa la conformación energéticamente más favorable de la proteína, por ej: su estado nativo.
Una característica primaria de la interfaz de usuario (GUI) es el protector de pantalla que muestra el progreso actual durante el proceso Plegamiento de proteínas. En la parte superior izquierda del protector de pantalla actual, la proteína objetivo es visualizada adoptando diferentes formas (conformaciones) en su búsqueda por la estructura de energía mínima. Inmediatamente a la derecha se gráfica la más recientemente aceptada. En el extremo superior derecho se gráfica la menor conformación de energía del señuelo actual, a continuación se ha determinado cual es la estructura de la proteína correcta o nativa, en el protector de pantalla se han incluido tres gráficos. Hacia el centro se visualiza un gráfico del modelo de energía libre, que fluctúa a medida que el modelo aceptado cambia. Un gráfico de la raíz cuadrada media (RMSD) que mide cuan estructuralmente similar es el modelo aceptado al modelo nativo, es mostrado más alejado a la derecha. A la derecha del gráfico de energía aceptado y abajo del gráfico RMSD, los resultados de estas dos funciones son usados para producir un gráfico de energía contra RMSD para que el modelo sea progresivamente refinado.[11]
Como todos los proyectos Boinc este corre en modo background en la computadora del usuario usando recursos ociosos de procesamiento. Cuando los recursos son requeridos por otras aplicaciones Rosetta@home los libera para no perjudicar el rendimiento del equipo.
El usuario puede configurar el porcentaje que puede usar Rosetta@home del procesador para minimizar el consumo de energía y evitar sobrecalentamientos por el uso a límite del mismo.
También puede ser configurado el horario en que Rosetta@home se ejecutará así como otras preferencias desde un menú de configuraciones del usuario.
La versión original de Rosetta@home fue escrita en Fortran pero más tarde fue reescrita en C++ para permitir un desempeño más ágil. Esta nueva versión orientada a objetos fue lanzada el 8 de febrero de 2008. El desarrollo del código fue hecho por Rosetta Commons.[12] El software tiene licencia libre para la comunidad académica y disponible para las compañías farmacéuticas mediante el pago de una licencia.
Importancia del proyecto
Con la proliferación de proyectos de secuenciación de genomas, los científicos pueden inferir la secuencia de aminoácidos, o estructura primaria, de varias proteínas que acarrean funciones dentro de la celda. Para entender mejor la función de una proteína y ayudar en el diseño de drogas racionales, los científicos necesitan conocer la estructura terciaria tridimensional de la proteína. Las estructuras de las proteínas 3D están siendo actualmente a través de Cristalografía de rayos X o espectroscopia de resonancia magnética nuclear. El proceso es lento (lleva semanas o aun meses mostrar como cristalizar una proteína por primera vez) y tiene un alto costo (alrededor de 100000 U$S por proteína).[13] Desafortunadamente el valor en que cada nueva secuencia es descubierta excede lejos el valor de la determinación de la estructura, hay más de 7400000 secuencias de proteínas disponibles en la base de datos de proteínas no redundante NCBI y menos de 52000 estructuras de proteínas 3D han sido resueltas y depositadas en el Banco de Datos de proteínas, el repositorio principal para la información estructural de las proteínas.[14] Uno de los objetivos principales de Rosetta@home es predecir la estructura de la proteína con la misma precisión que los métodos existentes, pero que requiera mucho menos tiempo y dinero. Rosetta@home también desarrolla métodos para determinar la estructura de acoplamiento de las proteínas membrana (por ej: GPCR) que son excepcionalmente difícil de analizar con Cristalográfica de rayos X y espectroscopia NMR que todavía representa la mayoría de los objetivos para las drogas modernas.[15]
El progreso de la predicción de la estructura de la proteína es evaluada semestralmente en el experimento CASP ( Evaluación crítica de las técnicas de predicción de estructura de proteínas)[16] en el cual los investigadores a lo largo del mundo intentan derivar una estructura de una proteína desde la secuencia de aminoácidos de la misma. Los grupos altamente calificados en este experimento algunas veces competitivo son considerados estándares de facto en la técnica de predicción de la estructura de la proteína. Rosetta, el programa en el cual está basado Roseta@home ha sido usado desde CASP5 en 2002. En el experimento CASP6 de 2004 Rosetta hizo historia siendo el primero en producir un cierre a una resolución de nivel atómico, la predicción de la estructura de la proteína ab initio en su modelo presentado en el objetivo CASP T0281.[17] El modelamiento ab initio es considerado una categoría especialmente difícil de la predicción de la estructura de la proteína, porque no usa información desde el alineamiento estructural y debe contar con información del alineamiento múltiple de secuencia y modelamiento de interacciones físicas dentro de la proteína. Rosetta@home está siendo usado en CASP desde el 2006, y estuvo entre los mejores predictores en todas las categorías en CASP7.[18] [19] [20] Estas predicciones de alta calidad fue posible gracias a la potencia de cómputos de los voluntarios.[21] El incremento de la potencia de computo permite a Rosetta@home tomar muestras de más regiones del espacio de configuración (las formas posibles que puede tener una proteína), que de acuerdo a la paradoja de Levinthal[22] se predice que sea de incremento exponencial con la longitud de la proteína.
Rosetta@home se usa también en predicción de acoplamiento proteína-proteína, que determina la estructura de múltiples complejos proteicos, o estructura cuaternaria. Este tipo de interacción de proteínas afectan varias funciones celulares, incluyendo vínculos antigen-anticuerpo e inhibidores de enzimas e importación y exportación celular. Determinar estas interacciones es crítico para el diseño de drogas, Rosetta es usado en el experimento denominado Evaluación crítica de Predicción de Interacciones (CAPRI), que evalúa el estado del campo de acople de la proteína similar a como CASP mide progresos en la predicción de la estructura de la proteína. La potencia de computo disponibles por los voluntarios de Rosetta@home ha sido citada como un factor mayor en la perfomance en CAPRI donde sus predicciones de acoplamiento han sido entre las más exactas y completas.[23]
En los primeros años de la década del 80, Rosetta fue usado para diseñar computacionalmente una proteína con una función nunca observada en la naturaleza.[24] Fue inspirado en un artículo de alto nivel publicado en 2004 que describía originalmente el diseño computacional de una proteína con actividad enzimática mejorada con respecto a su forma natural.[25] El paper desarrollado por el grupo de David Baker describe como puede ser hecha la proteína con los recursos disponibles por Rosetta@home y representa una importante demostración conceptual para este método de diseño.[24] Este tipo de diseño de proteínas podría tener aplicaciones futuras en descubrimiento de drogas, química verde y biorremediación.
Enfermedades abarcadas por la investigación
Como agregado a la predicción básica en predicción de estructuras de proteína, diseño y acople, Rosetta@home es también usado en investigaciones relacionadas con enfermedades.[26] Proyectos de investigación menores son mencionados en la revista Rosetta@home de David Baker.[27]
El mal de Alzheimer
Un componente del paquete de programas Rosetta, RosettaDesign, fue usado para predecir con precisión que región de proteínas "amyloidgenicas"[28] que son más comunes para hacer fibrilas tipo amiloydes. Tomando hexapéptidos (fragmentos de seis aminoácidos grandes) de una proteína y seleccionando el patrón de energía más bajo de una estructura similar a la de una fibrila conocida de forma de hexapéptido. RossetaDesign fue capaz de identificar péptidos dobles con probabilidades de formar tanto fibrilas como proteínas aleatorias.[29] Rosetta@home fue usado en el mismo estudio para predecir estructuras para amyloide beta, una proteína en forma de fibrila que supone causa el mal de Alzheimer. Los resultados preliminares, pero aún no publicados han producido una proteína diseñada con Rosetta que pueden prevenir la formación de fibrilas, sin embargo se desconoce si puede producir el mal.[30]
Ántrax
RosettaDock,[31] [32] [33] otro componente de Rosetta@home fue usado en conjunto con métodos experimentales para modelar interacciones entre 3 proteínas, factor letal (LF), factor edema (EF), y antígeno protectivo (PA)— que hacen la toxinas del ántrax, el modelo computacional predice con precisión los acoplamientos entre LF y PA, ayudando a establecer que dominios de las proteínas respectivas se involucran en el complejo LF-PA. Esta predicción fue eventualmente usada en investigaciones para mejorar las vacunas contra el ántrax.[34] [35]
Virus herpes simplex 1
RossetaDock fue también usada para modelar estaciones de acople entre un anticuerpo (inmunoglobina G) y una proteína superficial expresada por el virus del herpes simplex 1 (HSV-1) que sirve para degradar el anticuerpo antiviral. La proteína compleja predicha por Rosetta@home coincide con los modelos experimentales particularmente difíciles de entender. Las principales investigaciones concluyen que los métodos de acople tienen el potencial de abordar algunos de los problemas que la cristalografía de rayos X tienen con las interfaces de modelaje proteína-proteína.[36]
HIV
Una parte de la fundación se financia mediante una subvención de 19,4 millones de dólares realizada por la fundación Bill and Melinda Gates.[37] </ Rosetta@home ha sido usada para diseñar múltiples vacunas posibles para el virus del SIDA.[38] [39]
La malaria
En las investigaciones relacionadas con la iniciativa "Grandes desafíos en salud global",[40] Rosetta has also been used to computationally design novel homing endonuclease proteins, which could eradicate Anopheles gambiae or otherwise render the mosquito unable to transmit malaria.[41] Rosetta ha sido usada para diseñar proteínas "homing endonuclease" que podrían erradicar el Anopheles gambiae u otra clase de mosquito capaz de transmitir la malaria. Siendo capaz de modelar y alterar la interacciones entre los ADNs de las proteínas, como otros "homing endonuclease", dan a las proteínas computacionales métodos de diseño como Rosetta un papel importante en terapias genéticas (que incluye posibles tratamiento del cáncer).[26] [42]
Historia del desarrollo y sus divisiones
Fue una idea del laboratorio Baker de 1998 como una mejora ab initio a las predicciones de estructuras,[43] ha sido dividido en diferentes fuentes de desarrollo y distintos servicios. Más de 7 años después de la primera aparición, Rosetta@home fue distribuido en el mercado (y como oficial no beta), el 6 de octubre de 2005[7] Varios de los estudiantes graduados en investigadores fueron trasladados a otras universidades y centros de investigación, y mejoraron diferentes partes del proyecto Rosetta.
RosettaDesign
RosettaDesign es una mejora computacional al diseño de proteínas basado en Rosetta, comenzó en el 2000 con un estudio en rediseñar la via plegable de la proteína G.[44] En 2002 el diseño RosettaDesign fue usado en dieño TOP7, un aminoácido de longitud α / β que tiene una carpeta total.[45] La nueva conformación fue prevista por Rosetta dentro de una RMSD 1.2Å de la estructura determinada por la cristalografía de rayos X representando una predicción de estructura inusualmente exacta[46] Rosetta y RosettaDesign ganaron un reconocimiento generalizado siendo el primero en diseñar y predecir exactamente la estructura de la proteína novel de tal longitud como se refleja en el artículo del 2002 describiendo la mejora dual propuesta en la revista Science y citada por más de 240 artículos científicos.[47] [48] El producto visible de la investigación,TOP7, fue destacado como la "Molécula del Mes" del Banco de Datos de proteínas en octubre de 2006, una superposición de sus respectivos núcleos (residuos 60-79) con su estructura de cristal son también destacadas en el logo de Roseta@home.[49]
Brian Khuhlman, un ex posdoctorado asociado en el laboratorio de David Baker y ahora profesor asociado en la Universidad de Carolina del Norte, Chapel Hill ofrece Rosetta@Design como un servicio online.[50] [51]
RosettaDock
RosettaDock fue agregado al paquete de software durante el primer experimento CAPRI, la predicción de algoritmos para el acoplamiento proteína-proteína del laboratorio Baker.[52] En ese experimento, RosettaDock una deducción altamente exacta para el acoplamiento entre la exótocina Streptococcus pyogenes A y un canal-β receptor de células T, y una predicción medianamente exacta para un complejo entre el α-amylase de los porcinos y un anticuerpo de los camélidos. Mientras que el método Rosetta@home solamente tiene dos predicciones aceptablemente exactas de siete posibles, fue suficiente para clasificarlo entre los métodos de predicción siete al noveno en las primeras pruebas CAPRI.[52]
El desarrollo de RosettaDock divergió en dos divisiones para los encuentros CAPRI subsiguientes como Jeffrey Gray que estableció las bases para RossetaDock mientras la Universidad de Washington continuaba trabajando en el método en la Universidad John Hopkins.[53] Los miembros del laboratorio Baker además desarrollaron RosettaDock en la ausencia de Gray. Las dos versiones difieren ligeramente en el modelamiento de la cadena, selección de señuelos y otros aspectos. A pesar de estas diferencias, tanto los métodos Baker y Gray ejecutaron correctamente en la segunda prueba CAPRI, ubicándose quinto y séptimo sobre otros 30 grupos de predictores.[54] El servidor RosettaDock de Jefrey Gray esta disponible como un servicio de prediccion libre para uso no comercial.[55]
En octubre de 2006, RosettaDock fue integrado dentro de Rosetta@home. El método usado fue una fase de modelo de acoplamiento crudo y rápido usando solamente la columna vertebral de la proteína. Seguido por una fase de refinamiento entera y lenta del átomo completo que consistió en optimizar la orientación de las dos proteínas interactuantes entre sí, y la interacciones de la cadena en la interfase proteína-proteína, fueron optimizadas simultáneamente para encontrar la conformación de menor energia.[56] La muy incrementada potencia computacional ofrecida por la red Rosetta@home, en combinación con las representaciones de "capa de árbol" revisadas para la flexibilidad de la columna vertebral y un modelamiento cíclico, hicieron de RosettaDock sexto de 63 grupos de predicción en la tercer prueba CAPRI.[4] [23]
Robetta
El servidor Robetta es un servicio de predicción de estructuras de proteína automatizado ofrecido por el laboratorio Baker para modelamiento comercial comparativo ab initio.[57] Ha participado como un servidor de predicción automatizado en el experimento semestral CASP desde CASP5 en 2002, ejecutando entre los mejores en la categoría servidores de predicción automatizados.[58] Robetta ha competido en CASP6 y 7, donde lo hizo mejor que el promedio entre servidores automatizados y grupos predictores humanos.[20] [59] [60]
En la estructura de modelamiento de proteínas como en CASP6, Robetta primero busca la estructura homologa usando BLAST, PSI-BLAST y 3D-Jury, luego compara la secuencia destino en sus dominios individuales o independientemente en unidades plegables de proteínas, igualando las secuencias a familias estructurales en la base de datos Pfarm. Los dominios con estructuras homologas siguen un protocolo de modelo basado en plantillas (por ej: modelamiento de homologías). Aquí el programa de alineación del laboratorio Baker doméstico, K*Sync ,produce un grupo de secuencias homologas, y cada una de estas es modelada por el método Rosetta de novo para producir una semilla (estructura posible). La predicción de estructura final es seleccionada tomando el modelo de menor energía determinado por una función de energía de baja resolución. Para los dominios que no tienen una estructura homologa detectada, un protocolo de novo es seguido en el modelo de energía más baja desde un conjunto de semillas es seleccionada como la predicción final.[61]
Estas predicciones de dominio son conectadas juntas para investigar interacciones interdominios, de nivel terciaria entre las proteínas. Finalmente, las contribuciones en cadena son modeladas usando un protocolo para búsqueda conformacional MonteCarlo.
En CASP8, Robetta fue aumentada para usar un método de refinamiento de alta resolución del átomo total,[62] the absence of which was cited as the main cause for Robetta being less accurate than the Rosetta@home network in CASP7.[21] la falta que fue citada como la causa principal de que Robetta sea menos exacta que la red Rosetta@home en CASP7.
Foldit
El 9 de mayo de 2008, después que los usuarios sugirieron una versión interactiva del programa de computación distribuida, el laboratorio Baker publicamente distribuyo Foldit, un juego de predicción de estructura online basada en la plataforma Rosetta.[63] El 25 de septiembre de 2008 Foldit tiene cerca de 59000 usuarios registrados.[64] El juego le da al usuario un juego de controles (p. ej: agitar, meneo, reconstruir) para manipular la columna vertebral y la cadena de aminoácidos de la proteína destino en conformaciones energéticamente más favorables. Los usuarios pueden trabajar soluciones individualmente. Los usuarios pueden trabajar en soluciones individualmente como solistas o colectivamente como "evolvers", puntos procedentes sobre cada categoría que puedan mejorar sus predicciones de estructura.[65] Los usuarios pueden individualmente competir con otros usuarios en una especie de "duelo", en que el jugador con la estructura de menor energía después de 20 movimientos gana.
Comparación con proyectos de informática distribuida similares
Hay varios proyectos computacionales que han estudiado áreas similares la de Rosetta@home, pero difieren en sus aportes investigativos.
Folding@home
Todos los grandes proyectos de computación distribuida en el área de investigación de proteínas, Folging@home es el único que no usa la plataforma BOINC.[66] Tanto Rosetta@home y Folding@home investigan enfermades degenerativas de la proteína (por ej: el mal de Alzheimer) pero Folfing@home lo hace más exclusivamente.[67] En lugar de usar métodos basados en diseño o en estructura para predecir comportamientos de amiloydes, por ejemplo, Folding@home usa dinámica molecular para modelar cuantas proteínas plegaron (o potencialmente desplegaron, y "subsequently aggregate").[68] En otras palabras, el fuerte de Folding@home es modelar el proceso del plegado de la proteína, mientras que el fuerte de Rosseta@home es el diseño computacional de las proteínas y la predicción de la estructura y acople de la proteína. Los dos proyectos también difieren significativamente en su poder computacional y diversidad de hosts. Promediando acerca de 4.3 petaFLOPS (4300 teraFLOPS) con un host base que incluye la PlayStation 3 y unidades de procesamiento gráficas.[69] Folding@home tiene cerca de 55 veces la potencia computacional de Rosetta@home, que promedia 77 teraFLOPS con un host base consistente solamente en PC's basadas en CPU's.[2]
World Community Grid
Tanto la fase 1 como la fase 2 del proyecto de plegamiento del Proteoma Humano(HPF), un suproyecto del World Community Grid, han usado el programa Rosetta para hacer anotaciones estructurales y funcionales de varios genomas. Sin embargo Richard Bonneau, científico de cabecera que la usa ahora para crear bases de datos para biólogos estuvo activo en el desarrollo original de Rosetta en el laboratorio de David Backer mientras cursaba su doctorado.[70] Para más información sobre la relación entre HPF1,HPF2 y Rosetta@home puede ser encontrada en el sitio web del investigador.[71]
Predictor@home
Así como Rosetta@home, Predictor@home se especializa en predicción de estructuras de proteínas, Predictor@home también planea desarrollar nuevas áreas para su plataforma de computación distribuida para diseño y acople de proteínas (usando el paquete CHARMM para dinámica molecular) comparándolo con Rosetta@home.[72] Rosetta@home usa el programa Rosetta para su prediccion de estructura y Predictor@home usa la metodología dTASSER.[73]
Otros proyectos de computación distribuida relacionado con la proteína corriendo sobre BOINC son: QMC@home, Docking@home, SIMAP y TANPAKU. RALPH@home, el proyecto Rosetta@home que testea nuevas versiones de aplicaciones, unidades de trabajo, y actualizaciones antes de ser trasladadas a Rosetta@home.[74]
Contribuciones voluntarias
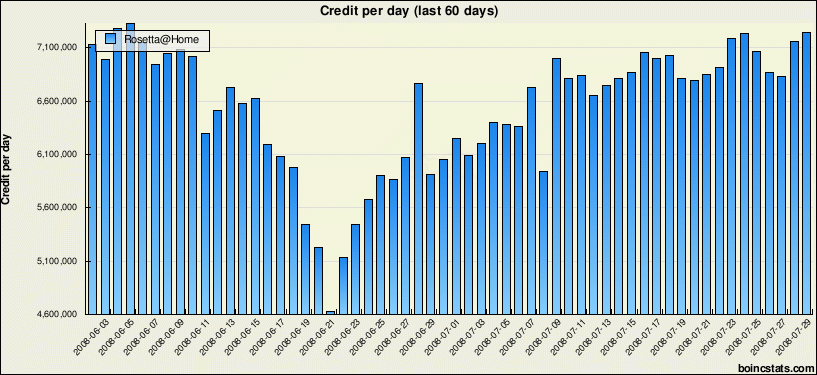 Gráfico de barras que muestra el crédito acumulativo diario para Rosetta@home sobre un periodo de 60 días, indicando su poder computacional durante el experimento CASP8.
Gráfico de barras que muestra el crédito acumulativo diario para Rosetta@home sobre un periodo de 60 días, indicando su poder computacional durante el experimento CASP8.
Rosetta@home depende del poder computacional donado por miembros del proyecto. Al 30 de noviembre de 2008, cerca de 47000 usuarios de 161 países fueron miembros activos de Rosetta@home, contribuyendo juntos con tiempo de procesamiento ocioso cerca de 86000 computadoras para una perfomance promedio combinada de cerca de 77 teraFLOPS.[2]
Los usuarios van obteniendo créditos BOINC a medida de sus contribuciones. El crédito obtenido por cada unidad de trabajo es el numero de semillas producida por tal unidad multiplicada por el crédito promedio de la semilla enviada por todos los hosts para esa unidad. Este sistema cliente fue diseñado para direccionar diferencias significantes entre créditos otorgados a usuarios con el cliente estándar BOINC y un cliente BOINC optimizado, una diferencia de créditos entre usuarios ejecutando Rosetta@home en Linux y en Windows.[75]
El monto de crédito obtenido por segundo de trabajo de CPU es menor para Rosetta@home que para los otros proyectos BOINC. A pesar de esta desventaja para los usuarios compitiendo en ranking, Rosetta@home esta quinto sobre otros 40 proyectos BOINC en términos de crédito total.[76]
Los usuarios de Rosetta@home que predicen estructuras de proteínas enviadas por el experimento CASP son dadas a conocer en publicaciones científicas en lo que respecta a los resultados.[77] Un "Usuario del dia" es elegido aleatoriamente cada día para estar en la página web
Los usuarios que predicen la estructura de energía más baja para una unidad de trabajo dada son destacados en la página web de Rosetta como el "Predictor del día" junto con cualquier otro miembro del equipo en el cual es miembro así como en la de usuarios que han hecho un perfil Rosetta@home.[78]
Referencias
- ↑ «Portfolio Highlight: Rosetta++ Software Suite». UW TechTransfer – Digital Ventures. Consultado el September 7 2008.
- ↑ a b c de Zutter W. «Rosetta@home: Credit overview». boincstats.com.
- ↑ «What is Rosetta@home?». Rosetta@home forums. University of Washington.
- ↑ a b Lensink MF, Méndez R, Wodak SJ (December de 2007). «Docking and scoring protein complexes: CAPRI 3rd Edition» Proteins. Vol. 69. n.º 4. pp. 704–18. DOI 10.1002/prot.21804. PMID 17918726.
- ↑ a b «Download BOINC client software». BOINC. University of California (2008).
- ↑ «Rosetta@home: Recommended System Requirements». Rosetta@home. University of Washington (2008).
- ↑ a b «Rosetta@home: News archive». Rosetta@home. University of Washington (2008).
- ↑ «Rosetta@home: FAQ (work in progress) (message 10910)». Rosetta@home forums. University of Washington (2006).
- ↑ http://en.wikipedia.org/wiki/Random_seed Definición en ingles de semilla aleatoria hasta que exista el articulo
- ↑ Kim DE (2005). «Rosetta@home: Random Seed (message 3155)». Rosetta@home forums. University of Washington.
- ↑ «Rosetta@home: Quick guide to Rosetta and its graphics». Rosetta@home. University of Washington (2007).
- ↑ «Rosetta Commons». RosettaCommons.org (2008).
- ↑ (2003) Bourne PE, Helge W (ed.). Structural Bioinformatics. Hoboken, NJ: Wiley-Liss. ISBN 978-0471201991.
- ↑ «Yearly Growth of Protein Structures». RCSB Protein Data Bank (2008).
- ↑ Baker D (2008). «Rosetta@home: David Baker's Rosetta@home journal (message 55893)». Rosetta@home forums. University of Washington.
- ↑ Artículo en wikipedia inglesa
- ↑ «Rosetta@home: Research Overview». Rosetta@home. University of Washington (2007).
- ↑ Kopp J, Bordoli L, Battey JN, Kiefer F, Schwede T (2007). «Assessment of CASP7 predictions for template-based modeling targets» Proteins. Vol. 69 Suppl 8. pp. 38–56. DOI 10.1002/prot.21753. PMID 17894352.
- ↑ Read RJ, Chavali G (2007). «Assessment of CASP7 predictions in the high accuracy template-based modeling category» Proteins. Vol. 69 Suppl 8. pp. 27–37. DOI 10.1002/prot.21662. PMID 17894351.
- ↑ a b Jauch R, Yeo HC, Kolatkar PR, Clarke ND (2007). «Assessment of CASP7 structure predictions for template free targets» Proteins. Vol. 69 Suppl 8. pp. 57–67. DOI 10.1002/prot.21771. PMID 17894330.
- ↑ a b Das R, Qian B, Raman S, et al (2007). «Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home» Proteins. Vol. 69 Suppl 8. pp. 118–28. DOI 10.1002/prot.21636. PMID 17894356.
- ↑ La paradoja de Levinthal, artículo en wikipedia inglesa
- ↑ a b Wang C, Schueler-Furman O, Andre I, et al (December de 2007). «RosettaDock in CAPRI rounds 6-12» Proteins. Vol. 69. n.º 4. pp. 758–63. DOI 10.1002/prot.21684. PMID 17671979.
- ↑ a b Jiang L, Althoff EA, Clemente FR, et al (March de 2008). «De novo computational design of retro-aldol enzymes» Science (New York, N.Y.). Vol. 319. n.º 5868. pp. 1387–91. DOI 10.1126/science.1152692. PMID 18323453.
- ↑ Hayden EC (February 13, 2008). «Protein prize up for grabs after retraction» Nature. DOI 10.1038/news.2008.569. Consultado en 2008.
- ↑ a b «Disease Related Research». Rosetta@home. University of Washington (2008).
- ↑ Baker D (2008). «Rosetta@home: David Baker's Rosetta@home journal». Rosetta@home forums. University of Washington.
- ↑ [en.wikipedia.org/Amyloid]
- ↑ Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D (March de 2006). «The 3D profile method for identifying fibril-forming segments of proteins» Proceedings of the National Academy of Sciences of the United States of America. Vol. 103. n.º 11. pp. 4074–8. PMC 1449648. DOI 10.1073/pnas.0511295103. PMID 16537487.
- ↑ Baker D. «Rosetta@home forum: Publications on R@H's Alzheimer's work? (message 54681)». Rosetta@home forums. University of Washington.
- ↑ Wang C, Schueler-Furman O, Baker D (May de 2005). «Improved side-chain modeling for protein-protein docking» Protein science : a publication of the Protein Society. Vol. 14. n.º 5. pp. 1328–39. PMC 2253276. DOI 10.1110/ps.041222905. PMID 15802647.
- ↑ Gray JJ, Moughon S, Wang C, et al (August de 2003). «Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations» Journal of molecular biology. Vol. 331. n.º 1. pp. 281–99. PMID 12875852.
- ↑ Schueler-Furman O, Wang C, Baker D (August de 2005). «Progress in protein-protein docking: atomic resolution predictions in the CAPRI experiment using RosettaDock with an improved treatment of side-chain flexibility» Proteins. Vol. 60. n.º 2. pp. 187–94. DOI 10.1002/prot.20556. PMID 15981249.
- ↑ Lacy DB, Lin HC, Melnyk RA, et al (November de 2005). «A model of anthrax toxin lethal factor bound to protective antigen» Proceedings of the National Academy of Sciences of the United States of America. Vol. 102. n.º 45. pp. 16409–14. PMC 1283467. DOI 10.1073/pnas.0508259102. PMID 16251269.
- ↑ Albrecht MT, Li H, Williamson ED, et al (November de 2007). «Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax» Infection and immunity. Vol. 75. n.º 11. pp. 5425–33. PMC 2168292. DOI 10.1128/IAI.00261-07. PMID 17646360.
- ↑ Sprague ER, Wang C, Baker D, Bjorkman PJ (June de 2006). «Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging» PLoS biology. Vol. 4. n.º 6. pp. e148. PMC 1450327. DOI 10.1371/journal.pbio.0040148. PMID 16646632.
- ↑ Paulson, Tom. «Gates Foundation awards $287 million for HIV vaccine research», Seattle Post-Intelligencer, July 19, 2006.
- ↑ Liu Y et al (2007). «Development of IgG1 b12 scaffolds and HIV-1 env-based outer domain immunogens capable of eliciting and detecting IgG1 b12-like antibodies» (PDF). Global HIV Vaccine Enterprise.
- ↑ Baker D. «David Baker's Rosetta@home journal archives (message 40756)». Rosetta@home forums. University of Washington.
- ↑ «Homing Endonuclease Genes: New Tools for Mosquito Population Engineering and Control». Grand Challenges in Global Health.
- ↑ Windbichler N, Papathanos PA, Catteruccia F, Ranson H, Burt A, Crisanti A (2007). «Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos» Nucleic acids research. Vol. 35. n.º 17. pp. 5922–33. PMC 2034484. DOI 10.1093/nar/gkm632. PMID 17726053.
- ↑ Ashworth J, Havranek JJ, Duarte CM, et al (June de 2006). «Computational redesign of endonuclease DNA binding and cleavage specificity» Nature. Vol. 441. n.º 7093. pp. 656–9. DOI 10.1038/nature04818. PMID 16738662.
- ↑ Simons KT, Bonneau R, Ruczinski I, Baker D (1999). «Ab initio protein structure prediction of CASP III targets using ROSETTA» Proteins. Vol. Suppl 3. pp. 171–6. PMID 10526365.
- ↑ Nauli S, Kuhlman B, Baker D (July de 2001). «Computer-based redesign of a protein folding pathway» Nature structural biology. Vol. 8. n.º 7. pp. 602–5. DOI 10.1038/89638. PMID 11427890.
- ↑ [1]
- ↑ Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D (November de 2003). «Design of a novel globular protein fold with atomic-level accuracy» Science (New York, N.Y.). Vol. 302. n.º 5649. pp. 1364–8. DOI 10.1126/science.1089427. PMID 14631033.
- ↑ Jones DT (November de 2003). «Structural biology. Learning to speak the language of proteins» Science (New York, N.Y.). Vol. 302. n.º 5649. pp. 1347–8. DOI 10.1126/science.1092492. PMID 14631028.
- ↑ von Grotthuss M, Wyrwicz LS, Pas J, Rychlewski L (June de 2004). «Predicting protein structures accurately» Science (New York, N.Y.). Vol. 304. n.º 5677. pp. 1597–9; author reply 1597–9. DOI 10.1126/science.304.5677.1597b. PMID 15192202.
- ↑ «Articles citing: Kuhlman et al (2003) 'Design of a novel globular protein fold with atomic-level accuracy'». ISI Web of Science.
- ↑ «Kuhlman laboratory homepage». Kuhlman Laboratory. University of North Carolina.
- ↑ «RosettaDesign web server». Kuhlman Laboratory. University of North Carolina.
- ↑ a b Gray JJ, Moughon SE, Kortemme T, et al (July de 2003). «Protein-protein docking predictions for the CAPRI experiment» Proteins. Vol. 52. n.º 1. pp. 118–22. DOI 10.1002/prot.10384. PMID 12784377.
- ↑ Daily MD, Masica D, Sivasubramanian A, Somarouthu S, Gray JJ (2005). «CAPRI rounds 3-5 reveal promising successes and future challenges for RosettaDock» Proteins. Vol. 60. n.º 2. pp. 181–86. DOI 10.1002/prot.20555. PMID 15981262.
- ↑ Méndez R, Leplae R, Lensink MF, Wodak SJ (2005). «Assessment of CAPRI predictions in rounds 3-5 shows progress in docking procedures» Proteins. Vol. 60. n.º 2. pp. 150–69. DOI 10.1002/prot.20551. PMID 15981261.
- ↑ «RosettaDock server». Gray laboratory. Johns Hopkins University.
- ↑ «Protein-protein docking at Rosetta@home». Rosetta@home forums. University of Washington.
- ↑ «Robetta web server». Baker laboratory. University of Washington.
- ↑ Aloy P, Stark A, Hadley C, Russell RB (2003). «Predictions without templates: new folds, secondary structure, and contacts in CASP5» Proteins. Vol. 53 Suppl 6. pp. 436–56. DOI 10.1002/prot.10546. PMID 14579333.
- ↑ Tress M, Ezkurdia I, Graña O, López G, Valencia A (2005). «Assessment of predictions submitted for the CASP6 comparative modeling category» Proteins. Vol. 61 Suppl 7. pp. 27–45. DOI 10.1002/prot.20720. PMID 16187345.
- ↑ Battey JN, Kopp J, Bordoli L, Read RJ, Clarke ND, Schwede T (2007). «Automated server predictions in CASP7» Proteins. Vol. 69 Suppl 8. pp. 68–82. DOI 10.1002/prot.21761. PMID 17894354.
- ↑ Chivian D, Kim DE, Malmström L, Schonbrun J, Rohl CA, Baker D (2005). «Prediction of CASP6 structures using automated Robetta protocols» Proteins. Vol. 61 Suppl 7. pp. 157–66. DOI 10.1002/prot.20733. PMID 16187358.
- ↑ Baker D. «David Baker's Rosetta@home journal, message 52902». Rosetta@home forums. University of Washington.
- ↑ Baker D. «David Baker's Rosetta@home journal (message 52963)». Rosetta@home forums. University of Washington.
- ↑ «Foldit forums: How many users does Foldit have? Etc. (message 2)». University of Washington.
- ↑ «Foldit: Frequently Asked Questions». fold.it. University of Washington.
- ↑ «Project list - BOINC». University of California.
- ↑ «Folding@home - FAQ-Diseases». Stanford University (2007).
- ↑ «Folding@home - About». Stanford University (2008).
- ↑ «Client statistics by OS». Stanford University.
- ↑ «List of Richard Bonneau's publications». Bonneau Lab, New York University.
- ↑ Bonneau R. «World Community Grid Message Board Posts». Bonneau Lab, New York University.
- ↑ «Predictor@home: Developing new application areas for P@H». The Brooks Research Group.
- ↑ Carrillo-Tripp M (2007). «dTASSER». The Scripps Research Institute.
- ↑ «RALPH@home website». RALPH@home forums. University of Washington.
- ↑ «Rosetta@home: The new credit system explained». Rosetta@home forums. University of Washington (2006).
- ↑ «BOINCstats: Project Credit Comparison». boincstats.com (2008).
- ↑ Das R, Qian B, Raman S, et al (2007). «Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home» Proteins. Vol. 69 Suppl 8. pp. 118–28. DOI 10.1002/prot.21636. PMID 17894356.
- ↑ «Rosetta@home: Protein Folding, Design, and Docking». Rosetta@home. University of Washington (2008).
Enlaces externos
- La revista Rosetta@home de David Baker
- Incluye una descripción general de la platforma así como una guía para la instalación de BOINC con el agregado de Rosetta@home
- BOINCstats – Rosetta@home Estadísticas detalladas de las contribuciones
- RALPH@home Website for Rosetta@home alpha testing project
- Rosetta@home Página web del proyecto
- Video en youtube con la descripción general hecha por David Baker
- Rosetta Commons Plataforma académica colaborativa para el desarrollo de Rosetta
Servicios de Rosetta online
- Robetta Servidor de predicción de la estructura de la proteína
- RosettaDesign Servidor de diseño de la proteína
- RosettaDock, Servidor de estación de acoplamiento proteína-proteína
- Todo o parte de este artículo fue creado a partir de la traducción del artículo Rosetta@home de la Wikipedia en inglés, bajo licencia Creative Commons Compartir Igual 3.0. y GFDL.
Categorías: Bioinformática | Computación distribuida
Wikimedia foundation. 2010.