- Cistinuria
-
Cistinuria 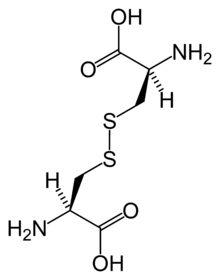
Estructura química de la cistina formada por L-cisteína (en condiciones biológicas)Clasificación y recursos externos CIE-10 E72.0 CIE-9 270.0 OMIM 220100 IIER 42 DiseasesDB 3339 MedlinePlus 000346 MeSH D003555  Aviso médico
Aviso médicoLa cistinuria es una enfermedad hereditaria con un patrón de herencia autosómica recesiva,[1] con una incidencia estimada de un caso por cada 7.000 nacidos vivos, que ocasiona una elevada excreción urinaria de aminoácidos dibásicos como cistina, lisina, arginina, y ornitina.[2]
 Estructura química de la cistina.
Estructura química de la cistina.
Contenido
Historia
Fue descrita a principio del siglo XIX e incluída en 1908 entre los primeros errores congénitos del metabolismo conocidos por Sir Archibald Edward Garrod.
En 1810, Wollaston, observó unos cálculos diferentes a los usuales, compuestos por placas hexagonales y solubles en álcali y pensó, erróneamente, que eran exclusivamente de origen vesical. Marcet, en 1817, mostró que este tipo de litiasis podía presentarse en el riñón.[3]
Poco tiempo después, en 1824, Stromeyer identificó en la orina de pacientes afectos los típicos cristales que han servido – y siguen sirviendo – para el diagnóstico de la enfermedad.[4] Hasta 1947, no se comprobó que estaba asociada a una excreción urinaria excesiva de tres aminoácidos: arginina, lisina, y ornitina.[5] [6] La hipótesis de que era causada por un defecto en la reabsorción renal e intestinal de estos aminoácidos y la cistina, formulada por Dent y Rose en 1951, fue confirmada en 1961 por Milne. La primera descripción del patrón hereditario fue realiza por Harris en 1955.[7]
Bases genéticas
Se trata de una enfermedad hereditaria en la que, como consecuencia de mutaciones en los genes SLC3A1 (ubicado en el cromosoma 2, 2p16.3-21) y SLC7A9 (situado en el cromosoma 19, 19q12-13), se ve afectado el transporte a nivel tubular renal y en algunos casos intestinal, de determinados aminoácidos dibásicos: arginina, lisina, ornitina, así como el de la cistina (dímero del aminoácido cisteína).[8]
Bases fisiopatológicas
En una persona sin cistinuria, los aminoácidos que llegan a los riñones en el torrente sanguíneo son filtrados a través del glomérulo renal y posteriormente reabsorbidos a la sangre por mecanismos de transporte activo. En adultos, el 99 % de la cistina filtrada es reabsorbida. Los aminoácidos dibásicos: cistina, arginina, lisina y ornitina, durante la reabsorción, atraviesan las células epiteliales del riñón, y vuelven a la sangre por medio de la arteria eferente en condiciones normales. Todavía, en pacientes con cistinuria, la cistina es filtrada, pero no es reabsorbida en el riñón. La mutación de los genes SLC3A1 y SLC7A9 provoca un defecto metabólico que afecta a las proteínas transportadoras de estos aminoácidos dibásicos y por esto no pueden atravesar la membrana de las células epiteliales del riñón, siguiendo el camino de la excreción. Este sistema de transporte se encuentra en la superficie luminal de las células epiteliales renales e intestinales. Perteneciente a los transportadores heteroméricos, está constituido por dos polipéptidos: una subunidad pesada y una ligera unidas por puente disulfuro. La subunidad pesada está formada por glicoproteínas N-glicosiladas tipo II de membrana, mientras que la subunidad ligera, lo está por proteínas no glicosiladas de membrana. La proteína perteneciente al primer grupo rBAT está implicada en un sistema de reabsorción de alta afinidad para la cistina en proximal de la nefrona y en el tracto intestinal.
Por tanto, podemos definir a la cistinuria como un defecto hereditario en el transporte de alta afinidad para la cistina, compartido con aminoácidos dibásicos, a través de células epiteliales del túbulo renal y enterocitos yeyunales.[8]
Síntomas
Los principales síntomas de esta enfermedad son el sangre en la orina (hematuria) y dolores en el costado o la espalda. Pueden también sentirse dolores en la pelvis, la ingle, los genitales o entre la parte superior del abdomen y la espalda.[9]
 Localización de los dolores provocadas por la Cistinuria.
Localización de los dolores provocadas por la Cistinuria.Diagnóstico
El diagnóstico definitivo se realiza según a la información aportada por el laboratorio mediante la determinación de la composición exacta de los cálculos, por constatación de la hipercistinuria o bien, mediante la caracterización del gen implicado.
El procedimiento diagnóstico más simple, aunque sólo demostrable en el 19-26 % de los pacientes homocigotos, es la identificación microscópica en el sedimento urinario de los típicos cristales hexagonales planos, ya sean aislados o bien formando maclas. Son más fácilmente observables en la primera orina de la mañana, ya que es más ácida y concentrada; en cualquier caso si acidificamos la orina con ácido acético conseguiremos la precipitación de cristales que no son visibles con orina fresca. El aspecto macroscópico que presentan los cálculos puros de cistina, observado mediante microscopía estereoscópica (lupa binocular), es de color miel-caramelo, céreo, brillante muy duro; de morfología y tamaño variable (aunque generalmente redondeada y de gran tamaño). Su estructura microcristalina se puede caracterizar mediante: microscopía diferencial de polarización, microscopía electrónica de barrido y microanálisis por energía dispersiva de rayos X, siendo diagnósticas las formas prismáticas hexagonales, generalmente en maclas.[10] El análisis genético, aunque definitivo en casos individuales, no parece ser de utilidad en el cribado de la cistinuria, dado la baja frecuencia de mutaciones (baja prevalencia de la enfermedad) y la variabilidad de éstas.[11] [12]
Tratamiento
El tratamiento de esta enfermedad consiste en facilitar la disminución de la concentración de cistina en la orina de estos pacientes, por lo que se recomienda beber más de 3 o 4 litros de agua al día. Además es importante alcalinizar la orina, con diversos preparados como el bicarbonato sódico, para aumentar la solubilidad de la cistina. El tratamiento invasivo es el clásico de las litiasis: cirugía abierta, nefrolitotomía percutánea (que también permite el uso de la litotricia ultrasónica o litoclastia con láser-holmio), citoscopía, endourología ureteral con litofragamentación directa con láser holmio, entre otros. En casos extremos, la combinación de infección y obstrucción puede conducir a una nefrectomía, en este caso procederá el trasplante renal ya que el nuevo riñón injertado no reproducirá el defecto en el transporte tubular de aminoácidos.[13]
Prevención
Una aproximación inicial al manejo del paciente cistinúrico requerirá un tratamiento preventivo con medidas como hidratación, moderada ingesta de sal (se aconseja una toma diaria inferior a 2 g), y alcalinización oral. La progresión de la enfermedad, hará procedente el tratamiento médico con los métodos anteriormente citados.[14] [15]
Véase también
Enlaces externos
- Fundación Internacional de Cistinuria (The International Cystinuria Foundation)[16]
Referencias
- ↑ Fjellstedt E, Harnevik L, Jeppsson JO, Tiselius HG, Söderkvist P, Denneberg T (diciembre 2003). «Excreción urinaria de cisteína total y el aminoacido dibásico arginina, lisina y ornitina en relación a hallazgos genéticos en pacientes con cistinuria tratado con compuestos sulfidrilos». Urological research 31 (6): pp. 417–425. doi:. PMID 14586528.
- ↑ http://www.ir.vhebron.net/easyweb_irvh/Activitats/Seminaris/delInsitut/Palacin/tabid/218/Default.aspx; 30 de octubre de 2009
- ↑ Marcet A. An essay on the chemical history and medical treatment of calculus disorders. En: Longman, Hurst, Rees, Orme y Brown editores. London, 1817. p. 79-88.
- ↑ Noehden GH. Scientific notices –chemistry,cystic oxide- communicated in a letter from Dr. Noehden to Mr. Childre. Annals of Philosophy 1824; 7: 146.
- ↑ Berzelius JJ. Calculus urinaries.Traité Chemistrie 1833; 7: 424-428.
- ↑ Yeh HL, Frankl W, Dunn MS, Parker P, Hughes B, Gyorgy P. The urinary excretion of amino acids by a cystinuric subject. Am J Med Sci 1947; 214: 507.
- ↑ Stein WH. Excretion of amino acids in cystinuria. Proc Soc Exp Biol Med 1951; 78: 705.
- ↑ a b Stein WH. Excretion of amino acids in cystinuria. Proc Soc Exp Biol Med 1951; 78: 705.
- ↑ Palacín M, Goodyer P, Nunes V, Gasparini P. Cystinuria. En: Scriver CR, Beaudet AL, Sly WS, Valle D, editores. The metabolic basis of inherited disease. 8ª ed. New York: McGraw-Hill; 2001; Vol III: p. 4909-4932
- ↑ Vilanova MA. Métodos habituales. En: Pinto B. Litiasis renal. 2ª ed. Barcelona: Masson-Salvat; 1993; 12-13.
- ↑ Guillén M, Corella D, Cabello ML, García AM, Sáiz C, González JI, Hernández-Yago J. Estudio genético y molecular de la cistinuria en la Comunidad Valenciana. Med Clin (Barc) 1999; 113: 321-326.
- ↑ Guillén M, Corella D, Cabello ML, Saiz C, Hernández-Yago J. Sensibilidad, especificidad y valor predictivo del análisis genético de variantes en el gen SLC3A1 aplicado al diagnóstico de cistinuria en la población española. Rev Clin Esp 2001; 201:256-259.
- ↑ Fjellstedt E, Denneberg T, Jeppsson JO, Tiselius HG. A comparison of the effects of potassium citrate and sodium bicarbonate nin the alkalinization of urine in homozygous cystinuria. Urol Res 2001; 29:295-302
- ↑ Assimos DG, Leslie SW, Ng C, Streem SB, Hart LJ. The impact of cystinuria on renal function. J Urol 2002; 168:27-30.
- ↑ Rousaud F, Gracia S, Palacín M, Nunes V, Millán F, Oliver A, Rousaud A. Cistinuria y litiasis renal de cistina. Estudio y enfoque terapéutico. Arch Esp de Urol 2001; 54: 989-996.
- ↑ «Cystinuria - International Cystinuria Foundation».
Categoría:- Enfermedades hereditarias

Wikimedia foundation. 2010.