- Talasemia
-
Talasemia 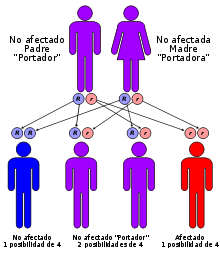
Herencia mendeliana autosómica recesiva: dos mutaciones de línea germinal (una de cada uno de los padres) para desarrollar la enfermedad; igualmente transmitida por hombres y mujeres.Clasificación y recursos externos CIE-10 D56. CIE-9 282.4 OMIM 141800 MedlinePlus Información de salud en la enciclopedia MedlinePlus PubMed Buscar en Medline mediante PubMed (en inglés) Sinónimos Anemia mediterránea. Anemia de Cooley.  Aviso médico
Aviso médico La talasemia es un grupo de anemias hemolíticas hereditarias en las que existe disminución de la síntesis de una o más de las cadenas polipeptídicas de la hemoglobina. Hay varios tipos genéticos con cuadros clínicos que van desde anomalías hematológicas difícilmente detectables hasta anemia severa y cuadros de enfermedad terminal.
Contenido
Prevalencia
Se estima que un 5% de la población mundial es portadora de un gen mutado para la hemoglobina (siendo más frecuente el ser portador de una talasemia que cualquier otra hemoglobinopatía). Unos 300.000 niños nacen cada año con síndromes talasémicos en todo el mundo.[1]
Descripción
La talasemia consiste en un grupo de enfermedades de amplio espectro. Estas van desde simples anormalidades asintomáticas en el hemograma hasta una severa y fatal anemia. La hemoglobina del adulto está compuesta por la unión de cuatro cadenas de polipéptidos: dos cadenas alfa (α) y dos cadenas beta (β). Hay dos copias del gen que produce la hemoglobina α (HBA1 y HBA2), y cada uno codifica una α-cadena, y ambos genes están localizados en el cromosoma 16. El gen que codifica las cadenas β (HBB) está localizado en el cromosoma 11.
En la α-talasemia gen HBA1[2] y HBA2 OMIM 141850, hay una deficiencia de síntesis de cadenas α. El resultado es un exceso de cadenas β que trasportan deficientemente el oxígeno, lo que conduce a bajas concentraciones de O2 (hipoxemia). Paralelamente, en la β-talasemia(OMIM 141900) hay una falta de cadenas beta, y el consiguiente el exceso de cadenas alfa puede formar agregados insolubles que se adhieren a la membrana de los eritrocitos, pudiendo causar la muerte de éstos y sus precursores, originando anemia de tipo hemolítico.
Causas moleculares de la enfermedad
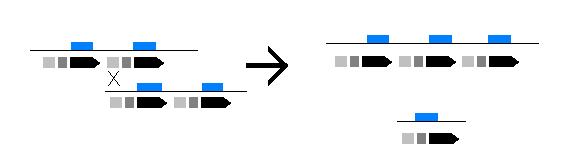
 Imagen de entrecruzamiento desequilibrado.
Imagen de entrecruzamiento desequilibrado.
Esta enfermedad está provocada por deleciones en uno o varios genes de los que componen los grupos de la α-globina y la β-globina. Según estas deleciones involucren más o menos genes el tipo de talasemia será más o menos grave.
Estas deleciones provocan la disminución en la producción de cadenas α o β, según el lugar de la deleción; la escasez de cadenas α se intenta compensar con un aumento de la producción de cadenas β, y viceversa, lo que da lugar a la formación de hemoglobinas inestables que provocan la destrucción de los glóbulos rojos y por lo tanto anemia.
A su vez las deleciones parecen ser el resultado de entrecruzamientos desequilibrados entre los segmentos duplicados presentes en la región de la agrupación.
En el caso de las β-talasemias además de la deleción del gen de la β-globina, también pueden darse por otras causas como:
- Mutaciones en el promotor que detienen o reducen su transcripción.
- Mutaciones en los sitios de corte y empalme (splicing) que impiden la eliminación de los intrones.
- Mutaciones en el sitio aceptor de poli-A que afectan al procesamiento del mRNA.
- Mutaciones de cambio en la pauta de lectura.
Síntomas
El defecto o deleción de un gen en la talasemia β causa una anemia hemolítica que oscila entre leve y moderada sin síntoma alguno. La deleción de dos genes ocasionan anemia más severa y la presencia de síntomas: debilidad, fatiga, dificultad respiratoria. En las variantes más graves, como la talasemia beta mayor, pueden aparecer ictericia, úlceras cutáneas, cálculos biliares y agrandamiento del bazo (que en ocasiones llega a ser enorme). La actividad excesiva de la médula ósea puede causar el ensanchamiento y el agrandamiento de algunos huesos, especialmente los de la cabeza y del rostro.
Los huesos largos tienden a debilitarse y fracturarse con gran facilidad. Los niños que padecen ciertas talasemias pueden crecer con más lentitud y llegar a la pubertad más tarde de lo normal. Como la absorción del hierro puede aumentar como respuesta a la anemia sumado al requerimiento de transfusiones de sangre frecuentes (las cuales suministran más hierro), es posible que se acumulen cantidades excesivas de hierro y se depositen en la musculatura del corazón, causando insuficiencia cardíaca.
Las talasemias son más difíciles de diagnosticar que otros trastornos de la hemoglobina. El análisis de una gota de sangre por electroforesis puede ser útil pero no concluyente, en especial en el caso de talasemia alfa. Por lo tanto, el diagnóstico se basa habitualmente en patrones hereditarios y en análisis especiales de hemoglobina. Por lo general, las personas que padecen talasemia no requieren tratamiento alguno, pero aquellas con variantes graves pueden requerir un trasplante de médula ósea. La terapia con genes se encuentra en fase de investigación.
Ventaja de sufrir alfa talasemia
La α-talasemia protege a los individuos que la portan frente a una enfermedad aún más grave, como es la malaria. La malaria o paludismo está producida por un parásito protista del género Plasmodium y es transmitida por un mosquito del género Anopheles. La protección frente a esta enfermedad por parte de los individuos que posee α-talasemia es debida a que Plasmodium sólo es capaz de parasitar a los eritrocitos sanos. Sin embargo, la sangre de alguien con este tipo de anemia presenta un número elevado de eritrocitos deformes por culpa de que la hemoglobina no está bien constituida y eso es esencial pues deja al parásito indefenso en la sangre permitiendo que nuestro sistema inmunitario acabe con él.
La ventaja del heterocigoto se produce cuando un alelo que es deletéreo en su forma homocigótica, resulta, en cambio ventajoso en su forma heterocigótica. Este fenómeno es lo que se llama polimorfismo equilibrado. Significa que la selección negativa del alelo en estado homocigótico se equilibra con la selección positiva a favor del alelo en el estado heterocigótico.
Debido a esto hay una alta frecuencia de talasemias, y en general de hemoglobinopatías en los países con malaria endémica, de modo que la distribución geográfica de la malaria se correlaciona con la de talasemias.
Clasificación
- α Talasemia rasgo (portador). Las mutaciones de la cadena α en el cromosoma 16 afecta a uno de los genes de un cromosoma causando una talasemia silenciosa (asintomática) caracterizada por algunas hemoglobinas con tres β y una α globina. Los portadores pueden tener mutaciones de la cadena α en dos cromosomas (16) afectando a un genes de cada cromosoma (o bien los dos genes de un solo cromosoma, estando normales los dos del cromosoma homólogo) causando una talasemia leve (puede ser asintomática) caracterizada por hemoglobinas con tres β y una α globina. Debido a que existen suficientes genes sin mutación en la cadena α la mayoría de las moléculas de hemoglobina tienen las respectivas dos cadenas α y dos β. La mayoría de los portadores de la α talasemia no lo saben y se descubre con análisis de ADN y biología molecular.[3]
- α Talasemia grave (Hemoglobina H). Las mutaciones de la cadena α en el cromosoma 16 afecta a tres de los genes (involucrando a ambos cromosomas homólogos) causando una talasemia grave caracterizada por la mayoría de las hemoglobinas con tres cadenas β y una α globina. Los afectados cursan con hemolisis intravascular causando anemia severa con los síntomas más graves.
- α Talasemia Mayor (Enfermedad de Bart). Las mutaciones de la cadena α en el cromosoma 16 afecta a los cuatro genes (involucrando a ambos cromosomas homólogos) causando un hidropesía fetal caracterizada por hemoglobinas con solamente cuatro cadenas γ (gamma) y es incompatible con la vida.[4]
- β+ Talasemia Menor (Minor). Las mutaciones de la cadena β en el cromosoma 11 afecta a uno de los genes causando una talasemia relativamente leve caracterizada por una hemoglobina con tres α y una β globina. Puede que no haya síntomas como puede que los síntomas sean intermedios entre leve y graves.[5]
- Enfermedad de la hemoglobina H La padecen aquellos individuos que sólo poseen una copia funcional del gen de la α-globina. Da lugar a una anemia moderada con inclusiones en los eritrocitos producidas por la hemoglobina H, la cual está formada por cuatro cadenas de β-globina, debida a las pocas que hay del otro tipo. Las manifestaciones clínicas son desde anemia leve a moderada, a veces puede llegar a provocar esplenomegalia.

- βº Talasemia Mayor (Major) o Anemia de Cooley.[6] Las mutaciones de la cadena β en el cromosoma 11 afectan a ambos genes causando la más grave de las talasemias caracterizada por la falta total de β globina. Cuatro cadenas α se combinan en defecto de las cadenas β formando una hemoglobina inestable que tiende a precipitarse en los glóbulos rojos causando daños en la membrana celular e incrementando la fragilidad del hematíe en cuestión. Por razón de la masiva hemolisis dirigida por el bazo, los síntomas son más graves: palidez, suceptibilidad a infecciones, fragilidad ósea, ictericia, depósitos de hierro en el hígado y corazón, y puede que no vivan mucho tiempo.
Diagnóstico
Las pruebas que se hacen para saber si un individuo padece cualquiera de los tipos de talasemia son análisis de sangre, que permiten ver la forma y la cantidad de glóbulos rojos en sangre. Otra forma de diagnosticar la enfermedad es por medio de estudios genéticos. Los cuales nos dan la información exacta del tipo de talasemia y su causa.
Actualmente, el análisis prenatal que se realiza mediante el muestreo de villus coriónico y la amniocentesis permite determinar la presencia o ausencia de talasemia en el feto, de esta forma si se detecta y es una forma grave puede ser tratada precozmente y que el individuo sobreviva.
Tratamiento
- Transfusiones de sangre: es el tratamiento que se utiliza para los tipos más graves de talasemia, como son la β-talasemia mayor. Los individuos que la poseen necesitan transfusiones regularmente, cada 2 ó 3 semanas, estas le ayudan a mantener la hemoglobina a niveles casi normales y a prevenir otras complicaciones, como la insuficiencia cardíaca y las deformidades óseas. Hay pacientes de otras talasemias menos graves que requieren esas transfusiones sanguíneas sólo ocasionalmente o porque desarrollan una enfermedad vira u otras infecciones, que puedan provocar que la anemia se vuelva más grave.
- Suplemento de ácido fólico: en el caso de que el individuo afectado sólo padezca anemia lo que se le suministra es ácido fólico, como para otros tipos de anemias.
- Uso de quelante de hierro: las transfusiones de sangre repetidas pueden resultar en la acumulación de hierro en el organismo. Esta acumulación puede ser perjudicial para el corazón, el hígado y otros órganos. Para prevenir estos daños, los individuos que se someten a transfusiones regularmente reciben un tratamiento con un tipo de medicamento llamado quelante de hierro, actualmente se utiliza la Deferoxamina. Este lo que hace es fijar el hierro y de esa manera se ayuda al organismo a deshacerse del exceso de este. La cantidad de hierro que tiene un individuo en sangre se mide por análisis de sangre, el problema es que no miden con demasiada precisión los niveles de hierro en el corazón e hígado, para eso hay que recurrir a una biopsia. Por eso la mayoría de los pacientes de talasemias graves que mueren es debido a ese acumulo de hierro.
- Trasplante de médula ósea: es tratamiento curativo. Este método es eficaz cuando el donante es perfectamente compatible desde el punto de vista genético, lo más compatibles son los miembros de la familia, como por ejemplo un hermano del individuo afectado. Con el trasplante de médula se logra curar al 85% de los individuos que consiguen un donante compatible. Sin embargo, sólo el 30% de los pacientes de talasemia consiguen un miembro de la familia que esté en condiciones de ser donante. El procedimiento es arriesgado y puede llevar a la muerte del paciente. Actualmente también se está utilizando para la cura de esta anemia y de otras la sangre del cordón umbilical de un hermano recién nacido y está siendo tan eficaz como el trasplante de médula ósea. Al igual que la médula ósea, la sangre del cordón posee células indiferenciadas, lo que se llama células madre que producen todas las demás células sanguíneas. Como ejemplo de este último método está el caso del niño sevillano que sufría beta talasemia mayor y que gracias a la sangre del cordón umbilical de su hermano recién nacido se ha curado. Ese hermano para que fuese donante totalmente compatible con el niño ha sido seleccionado genéticamente, siendo el primer bebé cuyo proceso de gestación y tratamiento genético ha tenido lugar íntegramente en España.[7]
Véase también: Hermano salvador- Terapia génica: por ser la beta-talasemia una enfermedad causada por un único gen ha sido objeto de estudio de la terapia génica. Los estudios con vectores retrovirales han demostrado su inestabilidad para trasnportar el gen de la beta-globina humana. Los mayores avances se han logrado empleando vectores lentivirales, con los cuales se consigue estabilizar la expresión de la beta-globina. Hasta el momento, los estudios en ratones han mostrado la corrección de la beta-talasemia en casos de media gravedad y mejoras parciales y variables cuando el fenotipo era grave. Mediante terapia génica in vitro empleando eritrocitos humanos de pacientes con beta-talasemias graves se ha conseguido restaurar una eritropoyesis funcional y revertir el elevado radio de apoptosis que caracteriza la beta-talasemia. El xenotrasplante de estas células en ratones obtuvo resultados positivos. No obstante, la terapia génica sigue en fases de ensayo clínico para esta enfermedad.
Puthenveetil, Geetha (2004). «Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector». Blood 104 (3445-3453).
Bibliografía
Peter Sudbery, 2004, Genética Molecular Humana, 2ª edición, Pearson
Nusboum, McInnes, Willard, 2004, Thompson & Thompson Genética en Medicina, 5º edición, Masson.
Véase también
- Beta-talasemia
- Alteraciones de los hematíes
- Enfermedad genética
- Anemia
- Sangre
Enlaces externos
- Asociación Española de lucha contra las hemoglobinopatías y Talasemias
- Fundación Argentina de Talasemia
- MedlinePlus Temas de salud: Talasemias.
- Manual Merck Capítulo 154: Anemias.
- http://www.saludinfantil.com/talasemia.htm
- http://cienciaaldia.wordpress.com/2009/05/08/dia-internacional-de-la-talasemia/
- http://www.publispain.com/revista/la-talasemia-o-anemia-mediterranea.htm
- http://www.ncbi.nlm.nih.gov/pubmed/20008179?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=8
- http://www.comtf.es/pediatria/Bol-2001-2/Hemoglobinopat%C3%ADas%20y%20Talasemias.pdf
- http://apps.who.int/gb/ebwha/pdf_files/EB118/B118_5-sp.pdf
Notas
- ↑ Organización Mundial de la Salud. Consejo Ejecutivo, reunión 11 de mayo de 2006. [1]
- ↑ OMIM 141800
- ↑ CLASIFICACIÓN AUTOMÁTICA DE RASGOS TALASÉMICOS Y OTRAS ANEMIAS MICROCÍTICAS E HIPOCROMICAS Javier Vicente y col. Universidad Politécnica de Valencia. Grupo de Bioingeniería, Electrónica y Telemedicina y Hospital Universitario Dr. Peset. Servicio de Hematología. Valencia España [2]
- ↑ Mauro Parra C. y col. ACTUALIZACIÓN EN EL DIAGNÓSTICO Y MANEJO DE ALTERACIONES HEMATOLÓGICAS DEL FETO Rev Chil Obstet Ginecol 2005; 70(1): 33-40 [3]
- ↑ HEMOGLOBINOPATÍAS Y TALASEMIAS J.J. Malcorra. BSCP Can Ped 2001; 25- no 2 [4]
- ↑ Yale Medical Group, the physicians of Yale University on the web. [5]
- ↑ Márquez, Noelia. Diario de Sevilla. Un trasplante pionero libra a un niño de padecer una enfermedad congénita. Sevilla, publicado el 14.03.2009. [6] Consultado el 27 de mayo de 2011
Categorías:- Enfermedades hereditarias
- Enfermedades hematológicas

Wikimedia foundation. 2010.