- Enfermedad de Niemann-Pick
-
Enfermedad de Niemann-Pick
Enfermedad de Niemann-Pick Descripción de la enfermedad
Clasificación y recursos externos Aviso médico
Aviso médico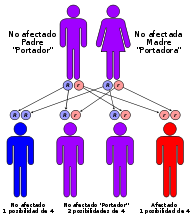
Herencia mendeliana autosómica recesiva: dos mutaciones de línea germinal (una de cada uno de los padres) para desarrollar la enfermedad; igualmente transmitida por hombres y mujeres. CIE-10 E75.23 CIE-9 272.7 OMIM 257200 Medline Buscar en Medline (en inglés) MedlinePlus enfermedad+de+niemann-pick Sinónimos Niemann-Pick.
Deficiencia de esfingomielinasa (enfermedad de Niemann-Pick tipo A). Síndrome de Crocker. Síndrome de Crocker-Farber. Enfermedad de Pick (Ludwig Pick).La enfermedad de Niemann-Pick es una enfermedad de almacenamiento lisosómico hereditaria autosómica recesiva, causada por mutaciones genéticas específicas, concretamente se trata de un déficit de la enzima esfingomielinasa, de la ruta de degradación de los esfingolípidos. Las cuatro formas de la enfermedad de Niemann-Pick se caracterizan por una acumulación de esfingomielina y colesterol en los lisosomas de las células, particularmente en las células de órganos importantes como el hígado y el bazo. Las tres formas más conocidas de la enfermedad son los tipos A, B y C.
El tipo A es poco frecuente, se caracteriza por su aparición temprana de la enfermedad, es muy severa, casi incompatibles con la vida, fallecen a los pocos meses y raramente sobreviven al primer año de vida.
El tipo B es otra manifestación de la enfermedad, el órgano afectado son los pulmones, son personas neurológicamente normales, suelen tener una longevidad aceptable.
El tipo C de la enfermedad de Niemann-Pick es de expresión neurológica y visceral. Se manifiesta con un marcado deterioro neurológico y en menor medida una afectación hepato-eplécnica. Tiene una forma de presentación infantil o precoz, con una esperanza de vida que no supera la primera década de vida. Y la presentación juvenil, con los mismos síntomas, pero con unas perspectivas de vida situada entre la segunda y tercera década de vida.
También existe el llamado tipo D:
El tipo D es muy parecido al tipo C en sus manifestaciones, considerándose una variante alélica de éste, en Nueva Escocia (Colorado).
No existe en el tipo C una relación clara con el déficit de esfingomielinasa, hay un origen genético causado por la anomalía en lo que se denominan, en términos de vanguardia, como genes reguladores. Pueden ser los causantes de esta defectuosa transcripción para la actuación de la enzima, se relaciona también a una proteína muy específica relacionada con la homeostasis del colesterol intracelular. Este depósito va a ir originando una alteración en las células deteriorándolas, deformándolas (células esponjosas) y terminando en muerte de estas. Los sustratos orgánicos donde van a repercutir esta lisis celular, primordialmente son por orden de importancia: cerebro, hígado y bazo. Al ser estos los órganos afectados, de ahí procederán las expresiones clínicas de esta enfermedad.
Los niños que la padecen mueren precozmente en los tres primeros años de vida. Las características de la enfermedad son infantilismo y trastornos del desarrollo. En la corteza cerebral aparecerán células hinchadas con la técnica de tinción de Baker, llamadas células xantomatosas. Otros signos son hepatoesplenomegalia, anemia, trastornos de la médula ósea, así como trastornos en los cartílagos de crecimiento de los huesos largos.
Contenido
Historia
La enfermedad de Niemann Pick se describió por primera vez en 1914 por el pediatra alemán Albert Niemann (nacido el 23 de febrero de 1880 en Berlín, lugar donde murió el 22 de marzo de 1921) en unos niños de origen judío (Askenazes), grupo étnico de centro y este de Europa; y en 1927 Ludwig Pick (nacido el 31 de agosto de 1868 y fallecido el 3 de febrero de 1944) ya como una entidad propia diferenciada de otras enfermedades, en un estudio tisular diferenciándola de la enfermedad de Gaucher. A Crocker le debemos la distinción, en 1961, de los cuatro tipos que hoy estudiamos (A, B, C y D). Finalmente citaremos que fue Brady quien aisló en 1966 la enzima lisosomal esfingomielinasa, cuyo déficit produce los tipos A y B. Los tipos C y D tienen como causa un defecto en el transporte intracelular del colesterol que se acumula en su forma libre sin esterificar.
Síntomas
Esta extraña y terrible enfermedad causa un progresivo deterioro en las funciones vitales, tales como:
- Pérdida de la capacidad de hablar.
- Pérdida paulatina de la habilidad de andar.
- Dificultad en la actividad intelectual.
- Dificultades para tragar alimentos.
- Insuficiencia respiratoria.
- Desconexión progresiva del medio que les rodea.
Diagnóstico
El diagnóstico de la enfermedad Niemann Pick se confirma con los estudios enzimáticos y con una biopsia en la piel del paciente, al mismo tiempo hay estudios moleculares que determina el tipo genético de la enfermedad.
Si un niño nace con algún tipo de Niemann-Pick (A, B, C) se debe a que los dos padres portan el gen anormal que lo produce aunque no tengan en ellos mismo síntomas de la enfermedad, esto es porque todos los tipos de la enfermedad son autosómicos recesivos.
Cuando los padres son portadores es más probable (50%) que el niño nazca portador de la enfermedad en vez de enfermo (25%), por esto es que se dan pocos casos de esta enfermedad rara.
Los exámenes médicos para detectar el portador en las familias todavía no es fiable. Pero para las mutaciones de los tipos A y B hay disponibles pruebas de ADN ya que se han estudiado ampliamente, sobre todo en la población judía asquenazí. En los pacientes de tipo C se han podido encontrar mutaciones del ADN y con esto, es posible encontrar al portador. En pocos centros médicos ya se dispone de un diagnóstico en el feto para el Niemann-Pick.
Muchos de los síntomas producidos por la enfermedad son comunes a otras enfermedades y esto dificulta el diagnóstico de certeza. Los médicos aconsejan que si se sospecha de la enfermedad en los tipos A y B, se realice una “medición de la actividad de la esfingomielinasa ácida en los glóbulos blancos”. Esto se consigue haciendo un análisis de sangre o una biopsia de células espumosas en la médula ósea. Esta prueba es ineficaz para detectar los portadores. Cuando la enfermedad es de tipo C, se diagnostica realizando una biopsia de piel, “cultivo celular en laboratorio y estudio posterior de la capacidad de las células aisladas para transportar y almacenar colesterol. Otras pruebas diagnósticas adicionales son la realización de un examen ocular con una lámpara de hendidura, la aspiración de la médula ósea y la biopsia del hígado.
Los científicos no pueden explicar por qué se producen tantas diferencias en la evolución de la enfermedad. Datos estadísticos puramente revelan que un niño con síntomas del tipo C antes de tener un año de edad no llega a alcanzar la edad escolar, pero si los presenta después de la escolarización, llegan a vivir hasta la mitad o el final de su adolescencia, muy pocos llegan hasta los 20 años. Aunque los tipos A y B se causan por lo mismo, una persona con el tipo A llega a la muerte sobre los 2 o 3 años, mientras que los de tipo B pueden llegar a la vida adulta.En toda España hay alrededor de 20 familias afectadas por el NP y realizan reuniones y conferencias sobre la enfermedad para discutir y analizar detenidamente los tratamientos preventivos posibles de la enfermedad que al catalogarse como “rara” y tener una prevalencia de uno entre un millón causa un interés nulo por parte de los laboratorios científicos para investigarla. De esta manera los familiares se ven obligados a sufragar los gastos con la ayuda de administraciones locales para la investigación.
Los más propensos a padecer el NP son los adolescentes y los niños aunque se sabe que puede atacar a cualquier edad, y la esperanza de vida es inferior a los 10 años en los niños y 30 en los jóvenes.
Evolución
El desarrollo de esta enfermedad se caracteriza por el paulatino deterioro de las funciones vitales del organismo. La enfermedad descubierta por los médicos Niemann y Pick es degenerativa y, por lo tanto, mortal en el 100% de los casos.
Tratamiento
Es recomendable para los pacientes tener una dieta baja en colesterol, pero ni esto ni los “medicamentos hipolipemiantes” detienen el progreso de la enfermedad. Pero muchos síntomas producidos por el tipo C (cataplexia y convulsiones) si se han podido frenar con medicamentos.
Aún no se ha encontrado un tratamiento eficaz para curar completamente esta enfermedad. Sin embargo, en la actualidad se están tratando de paliar las complicaciones que aparecen en el transcurso de la enfermedad mediante:
- Actividades fisioterapéuticas.
- Mejora en el ámbito nutricional.
- Tratamiento contra las crisis epilépticas originadas por el cúmulo de colesterol en el sistema nervioso.
- Tratamiento de [1]Zavesca con el fin de frenar la enfermedad.
Investigación
Desgraciadamente esta enfermedad no es objeto de investigación por los grandes programas de ciencia ni forma parte de la atención de los laboratorios químicos que no tienen mercado para sus productos, pues es una enfermedad minoritaria. Si bien, la Fundación Niemann Pick de España al frente de la Dras. M.J. Coll y Mercé Pineda no ha dejado de luchar en ningún momento. Además, y trabaja conjuntamente con otros organismos de distintos países con el mismo fin.
En la investigación para encontrar un tratamiento para los tipos A y B se ha progresado rápidamente desde los comienzos de los 90.Los expertos de la Universidad de Nueva York (Mount Sinai School of Medicine, ensayan desde hace más de 10 años terapias para combatir el Niemann-Pick y algunas de ellas (transplante de médula ósea, terapias genéticas y terapias de sustitución de enzimas) han tenido éxito dentro del laboratorio contra el NP de tipo B, pero ninguna contra el tipo A ni el C.
Años 2002-2004
Estudio de la expresión Fenotípica y de su relación con el genotipo en la enfermad de Niemann Pick Tipo C, en la población española.
- Objetivo: Prevención de la enfermedad. Posibles nuevas terapias.
Noviembre de 2003
Participación en el II Congreso médico mundial sobre la dicha enfermedad en Estados Unidos.
- Objetivo: Coordinar la labor y el esfuerzo científico médico entre profesionales e investigadores de Niemann Pick de España y el resto del mundo.
Mayo de 2004
Convenio suscrito con el laboratorio Actelion.
- Objetivo: Tratamiento experimental con un grupo de niños con Zavesca, con posibilidad de frenar la enfermedad.
Noviembre de 2004
Convenio de colaboración con la Universidad de Barcelona.
- Objetivo: Desarrollo de un estudio destinado a reducir la actividad del enzima glucosil ceramia sintasa, mediante la utilización de RNAs de interferencia pequeños como posible estrategia terapéutica para la enfermedad de Niemann Pick tipo C.
Años 2005-2007
Realización de un acuerdo de colaboración con la universidad de Barcelona (Departamento de Genética molecular).
2006-2008
Acuerdo con la fundación Clinic.
2008
Corrección del efecto de una mutación en fibroblastos de un paciente de Niemann Pick tipo C.
Investigaciones realizadas en el mundo
1) Se dispone de datos que afirman que se está desarrollando en Manchester un ensayo clínico (que aún está en fase inicial) que estudia a niños de entre 10 y 12 años.
2) En Ávila se presentó un ensayo que podría frenar los efectos de Niemann Pick. Contando con la colaboración de la Universidad de Barcelona, se realizó una investigación en la que se desarrolló de forma artificial la esfingomielinasa (que es la sustancia que hace que la grasa que se acumula en las células, en los enfermos de tipo B, se desprenda) y supondría un gran avance para los afectados.
3) Tras la llamada de atención de las familias afectadas, la Consejería de Sanidad se embarcó en un proyecto europeo “para fomentar las líneas de investigación de todas las enfermedades raras”. En 1999 fue aprobado un programa desde la UE de acción comunitaria sobre las enfermedades poco comunes. Se planteó la coordinación entre todas las administraciones para “complementar los esfuerzos de los distintos Estados miembros y maximizar el intercambio de información y experiencia, de modo que todos los Estados se encuentren asistidos para adoptar las medidas apropiadas cuando deban afrontar enfermedades con las que estén poco familiarizados” (Directiva 1.295/99/CE.
4) Desde el mes de mayo de 2002, se está probando en Inglaterra un medicamento (a manera de ensayo) con los niños que han desarrollado la enfermedad muy poco. Se trata de un remedio que quiere frenar el avance del NP pero se han detectado efectos secundarios peores que la enfermedad.
5) El proyecto "Signaling pathways leading to neuronal death in NPC: Finding new therapeutic targets", que se encarga de estudiar el tipo C, fue renovado por la fundación norteamericana “Ara Parseghian Medical Research Foundation”, tras presentar los resultados de la investigación en la “Scientific Conference on Nienmann-Pick Type C Disease” que se realizó a mediados de 2006 en Tucson (Arizona). Las doctoras Silvana Zanlugo, Alejandra Álvarez y Francisca Bronfman (inestigadora del Departamento de Gastroenterología de la Facultad de Medicina y directora del estudio, académica del Dep. de Biología Celular y Molecular; y profesora del Dep. de Ciencias Fisiológivas, respectivamente) son las que lideran el estudio.
- La primera etapa de la investigación, con duración de un año, se centró en entender las causas del NP.
- La parte más exitosa del proyecto es la que consistió en conseguir la mejora en el sistema locomotor de ratones afectados por el NP gracias a la aplicación de un inhibidor de una enzima.
- En la segunda etapa se buscará la profundización en el “estudio de las vías involucradas en la muerte neuronal que provoca esta afección” y a la “evaluación de posibles blancos terapéuticos que ayuden a detener su progresión”.
- El objetivo final es generar proteínas a partir de las que se puedan diseñar fármacos para prevenir la muerte de los pacientes.
6) Desde 2006 a 2008 se ha realizado un estudio con la ayuda de la “Beca de investigación Juan Girón” y en un convenio con la fundación “Clinic”. En la que durante el primer año “se puso en marcha de la técnica de Inmunoblot para detección de la proteína NPC1. El anticuerpo anti NPC”1, que es un policlonal, será suministrado por la Dra. Vanier de Lyon”; y durante el segundo, se realizó el mismo procedimiento pero con proteínas mutadas en lugar de la NPC1. Según los resultados obtenidos cabía la posibilidad de seguir estudiando en un tercer año.
7) En el año 2008 se produjo un gran anvace, pues se consiguió corregir el efecto de una mutación en fibroblastos en un paciente de tipo C, con la utilización de oligonucleótidos antisentido. Esto representa un paso para desarrollar una terapia para la enfermedad dirigida a los pacientes que tengan mutaciones de splicing y sen susceptibles al tratamiento con los oligonucleótidos antisentido. Este trabajo se realizó en la Universidad de Barcelona (Dep. de Genética de la Facultad de Biología) y con la dirección de la doctora Lluisa Vilageliu y el doctor Daniel Grinberg.
Estadísticas en España y el resto del mundo
Los consejos y el apoyo brindados desde el exterior nos llevaron a formar este grupo de trabajo, lugar desde donde nos mantenemos en permanente contacto con profesionales y laboratorios de E.E.U.U., España, Inglaterra, Francia, Israel, Japón y Suiza, lo cuál nos permite estar al tanto de las investigaciones que se realizando para lograr el tan ansiado objetivo que es la cura de la enfermedad. De acuerdo a las estadísticas en Argentina debería haber entre 20 y 50 casos, de los cuales en este momento no hay información centralizada, estaremos dentro o fuera de la media mundial, existirán o no estos casos, estarán o no bien diagnosticados. El hecho de ser una enfermedad de baja incidencia, sumado a que sus síntomas pueden ser asociados a otras patologías, más la desinformación existente, hacen que la enfermedad sea de muy difícil diagnóstico. EL equipo de trabajo está formado por un grupo humano solidario con esta realidad, amigos, médicos y profesionales de la salud abarcan las áreas de diagnóstico, neurología, nutrición, rehabilitación y psicología. Esperamos seguir sumando profesionales y anexar servicios para poder ayudar a quienes padecen esta cruel enfermedad.
Bibliografía
- A. Niemann: Ein unbekanntes Krankheitsbild. Jahrbuch für Kinderheilkunde, Berlín, N F, 1914. Volume 79: 1-10.
- L. Pick: Der Morbus Gaucher und die ihm ähnlichen Krankheiten (die lipoidzellige Splenohepatomegalie Typus Niemann und die diabetische Lipoidzellenhypoplasie der Milz). Ergebnisse der Inneren Medizin und Kinderheilkunde, Berlín, 1926, 29: 519-627.
- A. C. Crocker, S. Farber: Niemann-Pick disease: A review of eighteen patients. Medicine, Baltimore, 1958, 37: 1-95.
- Blanchete-Mackie EJ 2000. Intracellular Cholesterol Trafficking: role of the NPC1 Protein. Bba1486:171-183
- Carstea Ed Et Al. 1997. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277: 228-231
- Davies Jp Et Al 2000. Transmembrane molecular pump activity of Niemann-Pick C1 Protein. Science 290:2295-2297.
- Millat G Et Al. 1999. Niemann-Pick C1 Disease: the I1061T substitution is a frequent mutant allele in patients of western european descent and corelates with a classic juvenile phenotype. Am J hum genet 65:1321-1329
- Millat G Et Al., 2001. Niemann-Pick type C: spectrum of he1 mutations and genotype/ phenotype correlations in the NPC2 group. Am. J. Hum Genet., 69: on line
- Morris Ja Et Al 1999. The genomic organization and polymorphism analysis of the human Niemann-Pick C1 Gene. Bbrc 261:493-498
- Naureckiene S Et Al. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290:2298-2301
- Neufeld Eb Et Al.1999. The Niemann-Pick C1 protein residues in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J.Biol Chem 274:9627-9635
- Vanier Mt Et Al. 1996. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am J Hum Genet 58:118-125
- http://www.fnp.es/
- http://www.healthbasis.com/Spanish%20Health%20Illustrated%20Encyclopedia/5/001207.htm
- http://www.consumer.es/web/es/salud/investigacion_medica/2006/08/10/154507.php
- http://www.solociencia.com/noticias/0611/18133223.htm
- http://www.psiquiatria.com/noticias/pacientes_y_familiares/neuropsiquiatria/11042/
- http://www.jccm.es/revista/169/articulos169/salud_octubre.htm
- http://www.bio.puc.cl/bionoticias/bn_noticias_2007_001.htm
- http://www.consumer.es/web/es/salud/2003/11/10/90781.php
Véase también
- Anatomía patológica
- Enfermedad hereditaria
- Genes y enfermedades
- Consejo genético
- Prueba genética
Enlaces externos
- Genetics Home Reference (inglés)
Categorías: Enfermedades genéticas | Enfermedades metabólicas

Wikimedia foundation. 2010.