- Fenilcetonuria
-
Fenilcetonuria
Fenilcetonuria
Clasificación y recursos externos Aviso médico
Aviso médico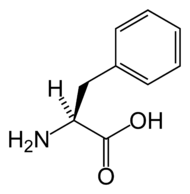
CIE-10 E70.0 CIE-9 270.1 OMIM 261600 DiseasesDB 9987 MedlinePlus 001166 eMedicine ped/1787 MeSH D010661 Sinónimos {{{sinónimos}}} La fenilcetonuria, es un tipo de hiperfenilalaninemia, también conocida como PKU, es una alteración del metabolismo en el que el organismo no puede metabolizar el aminoácido tirosina a partir de fenilalanina en el hígado. Esta enfermedad es genética y es provocada por la carencia de enzima fenilalanina hidroxilasa .
Antecedentes históricos
Esta enfermedad, de tipo oligofrénico, fue descrita por primera vez por el médico noruego Ivar Asbjørn Følling en 1934 cuando detectó la enfermedad en dos niños con retardo motor y mental cuya madre explicaba que los pequeños tenían un olor peculiar en la orina y el sudor. Éste síntoma específico recuerda al de los ratones o de establo y se nota en todos los afectados de fenilcetonuria. Al parecer es provocado por el ácido fenilacético eliminado vía renal y a través de los poros de la piel.
El doctor Følling creó un test con el que pudo detectar la acumulación del ácido fenilpirúvico por el color verde que produce al contacto con el cloruro férrico. Følling llamó primero a la enfermedad imbecillitas phenylpyruvica en virtud de que en las observaciones que hacía de la enfermedad notaba que ésta cursaba con un grave daño cerebral. Posteriormente y durante algún tiempo la enfermedad llevó su nombre: enfermedad de Følling. No fue sino hasta 3 años después de la primera descripción de la enfermedad que Lionel Penrose y Juda Hirsch Quastel la bautizaron como fenilcetonuria, que fue considerado un nombre más específico y con que se le conoce actualmente. Durante 25 años fue utilizado la prueba de reacción del cloruro férrico de Følling para la detección de la enfermedad en Europa y Estados Unidos.
En 1947, el doctor George A. Jervis, quien sería director del Instituto para la Investigación Básica de Problemas del Desarrollo, observó que cuando se administraba una dosis de fenilalanina a individuos sanos, se elevaba la tasa formación de tirosina, otro aminoácido. Sin embargo, cuando se suministraba la fenilalanina a individuos que padecían fenilcetonuria, no presentaban tal elevación. Los avances en el tratamiento no se iniciaron hasta 1953, año en que Jervis demostró que el defecto enzimático de la enfermedad involucraba la deficiencia de la enzima fenilalanina hidroxilasa que cataliza la reacción bioquímica que transforma la fenilalanina en tirosina. Ése mismo año, el doctor alemán Horst Bickel determinó que el exceso de fenilalanina en la dieta era la causa de la conducta hiperactiva y descontrolada de los pacientes con la enfermedad. Decidió, a petición de la madre de una de sus pacientes de dos años de edad, crear una dieta baja en fenilalanina, lo que mejoró sustancialmente la calidad de vida de la niña. A partir de las nuevas evidencias se publicaron varios trabajos en los que se demostraba la efectividad de las dietas bajas en fenilalanina, basadas en los hallazgos del doctor Jervis. Estos eventos llevaron a la creación de los programas de exámenes a recién nacidos que hoy en día se siguen utilizando.
En 1983, los doctores Li Chen y Savio L. C. Woo, ambos del Departamento de Medicina Celular y Genética, en la Escuela de Medicina Mount Sinai, aislaron e identificaron por primera vez el gen que codifica la elaboración de la enzima fenilalanina hidroxilasa, cuya carencia es responsable de la enfermedad.
En el transcurso de los años desde la primera descripción original de Følling hasta nuestros días, ha sido la fenilcetonuria, la única enfermedad que presenta deficiencia mental que puede ser evitada.
Etiología de la enfermedad
La fenilcetonuria tiene como rasgo principal la herencia genética autosómica recesiva, es decir, los padres son portadores de los genes defectuosos y al ser traspasados de ambos progenitores, la enfermedad se expresa en los descendientes.
La causa de la enfermedad es la carencia de la enzima fenilalanina hidroxilasa (FAOH) o de la dihidropterina reductasa (DHPR) (también llamada tirosina hidroxilasa). Ambas enzimas son responsables de la hidroxilación del aminoácido fenilalanina en la reacción que produce tirosina. Por ello, el defecto o falta de alguna de ellas determina un incremento de la concentración sanguínea de fenilalanina al impedirse su transformación en tirosina. También se aumenta la transaminación de la fenilalanina como vía metabólica alternativa, y asimismo se acumulan los metabolitos fenilpiruvato, fenilactato y fenilacetato. El defecto en la síntesis de FAOH se debe a una anomalía génica localizada en el cromosoma 12, y el de la DHPR en el cromosoma 4. Existen también formas de la enfermedad con déficits parciales.
El fenilpiruvato es un neurotori que afecta gravemente al cerebro durante el crecimiento y el desarrollo. Los efectos de su acumulación causan oligofrenia fenilpirúvica, caracterizada por un cociente intelectual inferior a 20. Los primeros meses de vida, los niños que padecen esta enfermedad parecen estar sanos. Entre los tres y los seis meses pierden el interés por el entorno, y al año se evidencia un retraso importante en su desarrollo. Los síntomas suelen ser retraso psicomotor, cuadros psicóticos de tipo autista, convulsiones, síndrome de West, convulsiones generalizadas y un eczema facial muy rebelde. Por lo general su desarrollo físico es bueno, tienden a tener el cabello más claro que sus hermanos, piel clara, y presentan un olor característico a paja mojada.
La técnica de Guthrie es la prueba que se utiliza para determinar esta enfermedad. Consiste en la detección de la fenilalanina mediante la inhibición que el metabolito β-2-tienialanina, derivado de la fenilalanina, produce sobre el crecimiento del Bacillus subtilis (cepa ATCC 60.51). El test de cribado tiene una sensibilidad y una especificidad cercanas al 99%.
Incidencia
Se estima que uno de cada 10,000 nacimientos puede presentar la enfermedad. Gracias a las pruebas de diagnóstico en neonatos es fácilmente detectable mediante los exámenes metabólicos habituales.
Los datos sobre la frecuencia de la fenilcetonuria en la población total de Europa y Norteamérica son exactamente iguales entre 2-6 casos por cada 100.000 habitantes. Donde es considerada más frecuente esta enfermedad es en los países del norte de Europa, donde son más corrientes los matrimonios entre vecinos y familiares.
El gen patológico es, al parecer, considerablemente más raro en las razas de color y los judíos que en los indogermanos. También es posible que el pequeño número de casos publicados sea debido a la falta de control rutinario de la orina. En China no se ha dado hasta ahora ningún caso de enfermedad de este tipo. También entre los europeos del sur, los indios de América del Norte y los gitanos la enfermedad aparece en muy pocas ocasiones. En Alemania viven actualmente unos 2.000 enfermos de este tipo.
Cuadro clínico
La enfermedad se manifiesta, por primera vez, algunas semanas después del nacimiento, iniciándose con una elevación en el plasma de la fenilalanina hasta un nivel 30 veces superior al normal y por la excreción de ácido fenilpirúvico por la orina. Transcurridos 6 meses se hace patente el retraso del desarrollo mental. La mayor parte de los pacientes son deficientes graves o profundos y en ocasiones se alcanza la deficiencia media.
El portador de esta anomalía, que nace tras un embarazo normal y sin complicaciones, se desarrolla durante los primeros meses casi siempre sin mostrar anormalidad ninguna. Sin embargo Partington encontró, casi en la mitad de los lactantes, la existencia de vómitos en los primeros meses de vida y en un tercio de ellos una irritabilidad inacostumbrada. En una proporción similar de casos, a los padres ya les había llamado la atención un desagradable olor del cuerpo del niño. Una parte de ellos mostró dermatosis eczematiformes durante el primer trimestre, 7 de 36 ya había tenido en el primer año de vida ataques convulsivos. A los 9 meses llama la atención el retraso en el desarrollo psicomotor.
Datos físicos
El desarrollo corporal cursa casi con normalidad. No obstante puede comprobarse cierta tendencia al enanismo, aunque también se han descrito casos con tallas superior a la frecuente. La dentición suele retrasarse hasta después del undécimo mes.
La gran mayoría de los enfermos muestran una piel clara, ojos azules, y color claro del pelo. Alrededor del 10% poseen cabellos oscuros. La pobreza de pigmentos llama más la atención en los pueblos de cabellos oscuros. La piel de los portadores además de ser clara es muy suave aterciopelada y muy sensible. En algunos enfermos se han observado eflorescencias papulosas en las caras de extensión de las extremidades y en la faz. En ciertos pacientes se puede encontrar también una tendencia a la acrocianosis.
Datos conductuales
Características clínicas raras
- Cifosis.
- Pies planos.
- Espina bifida.
- Sindactilia en los dedos de los pies.
- Bloqueo cardiaco intraventricular.
- Hipogenitalismo.
- Dermografismo.
- Sensibilidad a la luz.
- Hipersegmentación de las células neutrófilas de la sangre.
- Disminución de la tolerancia a la galactosa.
- Metabolismo basal ligeramente elevado.
Las etapas del desarrollo habitual, la edad en la que el niño se sienta y habla, a veces, se alcanzan a la edad normal, pero, de ordinario, se retrasa. En la edad límite en que debe esperarse que el niño normalmente realice estos actos, el 35% no puede andar y el 63% no puede hablar.
Estos niños, en general, tienen un peso y talla promedio por debajo del correspondiente a su edad. En la mitad de los casos tiene microcefalia y prominencia del maxilar.
Sus movimientos son lentos y patosos y a menudo suelen adoptar la posición de sastre. Las anomalías del tono muscular que contribuyen a estos cambios son de origen neurológico.2 de cada 3 pacientes tienen hiperreflexia tediciosa e hipercinesia sobreañadida estos últimos son voluntarios y muy variados.
Diagnóstico
El diagnóstico de la enfermedad se hace, prácticamente siempre, con ayuda de la positividad de la prueba del cloruro férrico en la orina. Existen casos raros en los que no se encuentra el ácido fenilpirúvico en la orina siempre, sólo cuando se ha expuesto al niño a una sobrecarga dietética o cuando tiene fiebre este aparece estos casos se consideran como defectos parciales
Biología hereditaria
 Gen autosómica recesiva. Modo de herencia entre dos portadores de un gen autosómica recesiva y monogénica.
Gen autosómica recesiva. Modo de herencia entre dos portadores de un gen autosómica recesiva y monogénica.
En una familia que presente un miembro con enfermedad manifiesta existe una probabilidad del 25% de que el próximo embarazo nazca de nuevo un portador de la anomalía. La transmisión se reparte por igual en los dos sexos.
En la mayoría de los casos los padres son normales. A menudo están emparentados entre sí, y raramente tienen otros parientes afectados. El nacimiento de niños normales de madres fenilcetonúricas establece el hecho de que el feto no recibe ningún daño irrecuperable por las anormalidades metabólicas de la madre.
Cuando en la familia del niño han existido portadores de la anomalía debe repetirse la prueba de Følling varias veces porque puede ocurrir que el ácido haya desaparecido al pasar unas horas. En los pañales el ácido se mantiene solo unas horas.
Inmediatamente después del nacimiento la prueba del cloruro férrico suele ser negativa. La positividad más precoz se inicia a los 14 días y la más tardía a los 35.
Tratamiento
Una terapéutica de la fenilcetonuria ( proteína de la leche ) es proporcionar solamente la cantidad de fenilanalina que se necesite para el crecimiento y la reparación de los tejidos. La reducción de la cantidad de fenilalanina, con dietas en las cuales las proteínas se sustituían por una costosa médula de aminoácidos puros sirve para que se mantengan en el cuerpo un nivel de concentración de fenilalanina tolerable.
Se ha intentado conseguir una reducción en la eliminación de ácido fenipirúvico por medio de la administración de dosis elevadas de fructosa. Las experiencias adquiridas con una dieta pobre en fenilalanina solo alcanzan hasta ahora para controlar durante pocos años la evolución de estos enfermos, pero ya puede asegurarse que con la iniciación precoz de esta terapéutica dietética se garantiza un desarrollo psíquico del niño aproximadamente normal.
Los ataque convulsivos, los síntomas neurológicos y la dermatosis desaparecen en los niños de más edad.
KUVAN ™ (SAPROPTERIN DIHIDROCLORURO) PARA PKU
Kuvan ™ (sapropterin dihydrohchloride), conocido anteriormente como Phenoptin ™, es el primero aprobado por la FDA para el tratamiento de fenilcetonuria (PKU), una enfermedad metabólica hereditaria causada por una deficiencia de la enzima fenilalanina hidroxilasa (PAH). Cantidades insuficientes o reducción de actividad de HAP se traduce en niveles elevados de fenilalanina (PHE) en la sangre. El mantenimiento de elevados niveles de Phe en sangre puede provocar graves daños neurológicos. Aprobado por la FDA en diciembre de 2007 Designado fármaco huérfano en los Estados Unidos y la Unión Europea; Fast Track ha concedido en los Estados Unidos BioMarin socio de Merck Serono, una división de Merck KGaA, Darmstadt, Alemania, presentó una solicitud de autorización de comercialización (MAA) a la Agencia Europea de Medicamentos (EMEA) en noviembre de 2007. Sapropterin dihidrocloruro, el ingrediente activo en Kuvan, es una forma sintética de 6R-BH4. 6R-BH4 es un cofactor esencial enzima que funciona en conjunción con HAP para metabolizar Phe.
Duración del tratamiento dietético
No se ha podido conseguir hasta ahora saber el tiempo durante el cual debe administrarse a estos enfermos la dieta pobre en fenilanina. Algunos autores defienden que la dieta debe seguirse durante toda la vida mientras que otros opinan que esta dieta puede suprimirse alrededor de los 10 años de vida.
La fenilalanina no puede suprimirse totalmente de la dieta, porque es un componente esencial de la alimentación. Aún en el mismo paciente la cantidad necesaria puede variar en diversas etapas de su vida.
Debe haber un equilibrio entre el aporte mayor y menor que sea lo suficiente en cantidad como para tener una máxima eficacia, disminuyendo el riesgo de toxicidad. Un aporte bajo en fenilalanina puede también ser fatal a la larga
Alimentos que contienen fenilalanina
- Leche materna
- Leche de vaca
- Huevos
- Pollo
- Cerdo
- Ternera
- Salmón
- Sardinas
- Gambas
- Caballa
- Mero
- Cereales
- Patatas
- Harina
- Espárragos
- Zanahorias
- Habas
- Arroz
- Coca Cola (de todos los tipos)
- Nutra sweet o Equal (estos son endulcorantes artificiales que tienen fenilalanina)
- Alimentos dieteticos con aspartamo
Hallazgos patológicos
La fenilcetonúria fuera del sistema nervioso central no produce alteraciones patológicas sobresalientes. Los cambios que han podido ser observados en el sistema nervioso central no explican la deficiencia mental. Hallazgos ocasionales han señalado alteraciones inespecíficas del hígado, alteraciones del tipo de la hipoplasia en la glándula pituitaria o en las gónadas.
La mayoría de las muertes ocurren en individuos jóvenes, después de una larga y extenuante enfermedad, que puede producir una gran variación de cambios patológicos sin relieve. Un hallazgo frecuente es que el peso del cerebro alcanza solo 2/3 del considerado como normal.
Procedimiento práctico
El sabor de todos los preparados que contienen proteínas tras la hidrólisis ácida es desagradable, por tanto su aporte proporciona considerables dificultades sobre todo en los periodos iniciales.
Después de haber planteado el diagnóstico se recurre a una alimentación totalmente libre de fenilalanina durante 10-20 días de esta forma se calcula el nivel exacto de proteínas que el paciente necesita. Este procedimiento se hace con todos los pacientes pues cada uno necesita cantidades especiales exactamente calculadas.
Pronóstico
Actualmente se está investigando con dietas pobres en fenilalanina en pacientes de hasta cuatro meses de vida desarrollándose los niños normalmente en todos los aspectos. En lactantes de mayor edad y sobre todo, en los niños pequeños, la mayoría de las veces las lesiones del sistema nervioso central son ya tan avanzadas e irreversibles que aún con una dieta especializada, solo puede conseguirse una escasa o moderada mejora del paciente con respecto a su capacidad psíquica.
Salvo raras excepciones, los pacientes que no son tratados terminan cayendo en retardo mental. Las posibilidades de supervivencia están acortadas sobre todo para los pacientes con retardo mental profundo.
Enlaces externos
- fenilcetonuria.es, web sobre fenilcetonuria para afectados e investigadores.
- Asociación Fenilcetonúrica de Galicia. PKU y OTM.
- http://www.medynet.com/usuarios/PrevInfad/Hipotiroidismo.htm#fenilcetonuria
- http://search.nacersano.org/cgi-bin/MsmGo.exe?grab_id=7&page_id=10878976&query=fenilcetonuria&hiword=fenilcetonuria+
- http://www.ultranet.com/~jkimball/BiologyPages/P/Phenylketonuria.html
- International PKU Board - Articles, Events, Recipes, etc.
- La fenilcetonuria: error innato del metabolismo
Categoría: Errores congénitos del metabolismo

Wikimedia foundation. 2010.