- Galactosemia
-
Galactosemia 
GalactosaClasificación y recursos externos CIE-10 E74.2 CIE-9 271.1 eMedicine ped/818 MeSH D005693  Aviso médico
Aviso médico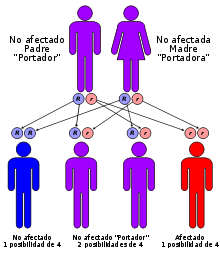 Herencia autosómica recesiva.
Herencia autosómica recesiva.
La galactosemia es una enfermedad hereditaria causada por una deficiencia enzimática y se manifiesta con incapacidad de utilizar el azúcar simple galactosa, lo cual provoca una acumulación de éste dentro del organismo, produciendo lesiones en el hígado y el sistema nervioso central.
Contenido
Concepto
La galactosemia es una enfermedad caracterizada por la incapacidad de metabolizar la galactosa en glucosa. La galactosa es un monosacárido obtenido principalmente de la hidrólisis de la lactosa contenida en la leche, aunque también puede estar presente en otros alimentos. La galactosa se absorbe en el intestino y principalmente se transforma en glucosa en el hígado.
Genética
La galactosemia sigue un patrón de herencia autosómico recesivo en sus tres tipologías. Generalmente los padres portadores asintomáticos tendrán un 25% de los hijos sanos, un 50% de los hijos portadores asintomáticos y un 25% de los hijos con galactosemia. Gene map locus: 9p13
Bioquímica
La metabolización de la galactosa a glucosa se realiza a través de la vía Leloir, la cual implica una serie de reacciones enzimáticas realizadas por diferentes enzimas:[1]
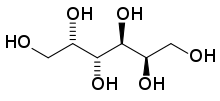 Galactitol.
Galactitol.Galactosa + ATP --- (Galactoquinasa) ---> Galactosa 1-fosfato + ADP.
Galactosa 1-fosfato + UDP-glucosa ---(Galactosa 1-fosfato uridiltransferasa) ---> Glucosa 1-fosfato + UDP-galactosa.
UDP-galactosa --- (UDP-galactosa 4-epimerasa) ---> UDP-glucosa.
Si la galactosa no es metabolizada por la vía Leloir puede metabolizarse por dos vías alternativas:En la primera la galactosa es reducida por una aldosa reductasa a galactitol, mientras que en la segunda la galactosa es oxidada por una galactosa deshidrogenasa a galactonato.[2]
Tipos
Las deficiencias de cada uno de estos enzimas producen los diferentes tipos de galactosemia:
Deficiencia de galactoquinasa (GALK). En este tipo de galactosemia la galactosa no puede ser fosforilada a galactosa 1-fosfato, por lo que se acumula en los tejidos y se metaboliza por las vías alternativas citadas anteriormente. Los genes mutados que codifican el enzima GALK se encuentran en los cromosomas 15 y 17. Su frecuencia estimada es de 1/150.000-1.000.000 de nacimientos.[2]
Deficiencia de UDP-galactosa 4-epimerasa (GALE). La reacción que transforma la UDP-galactosa en UDP-glucosa y viceversa no se realiza. El gen mutado que codifica el enzima GALE se encuentra en el cromosoma 1.[2] [3]
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Es el tipo más común y grave. También se conoce con el nombre de Galactosemia clásica. En este tipo de galactosemia la galactosa 1-fosfato no puede ser convertida a glucosa 1-fosfato. Se produce acumulación en los tejidos de galactosa y galactosa 1-fosfato. El gen que codifica el enzima GALT se encuentra en el cromosoma 9. Su frecuencia estimada es de 1/40.000-60.000 nacimientos.[2]
Síntomas
La sintomatología y su intensidad está determinada por el tipo de deficiencia enzimática que se presente.
Deficiencia de galactoquinasa (GALK). Únicamente se presenta la formación de cataratas debido a la acumulación de galactitol en el cristalino. No hay afectación de hígado, riñones o cerebro. Aumento de galactosa y galactitol en plasma y galactosuria.
Deficiencia de UDP-galactosa 4-epimerasa (GALE). Se pueden no mostrar síntomas o presentar síntomas parecidos a los de la galactosemia clásica. En ambos casos se produce una acumulación de UDP-Galactosa y Galactosa 1-fosfato.
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Se presentan letargo, rechazo al alimento y manifestaciones tóxicas generales, incluyendo vómitos y diarreas, pérdida de peso, ictericia, hepatomegalia, ascitis y la formación de cataratas entre otros debido a la acumulación de galactosa, galactitol y galactosa 1-fosfato en los tejidos. Aumento de galactosa y galactitol en plasma, galactosuria e hiperaminoaciduria.[2]
Diagnóstico
El díagnóstico se realiza por:[2]
- Cuantificación de galactosa y galactitol en plasma.
- Cuantificación de galactosa 1-fosfato, galactitol, galactonato y actividad enzimática GALK, GALE y GALT en glóbulos rojos.
- Presencia de sustancias reductoras en orina.
Posibles complicaciones
Las compliaciones más frecuente son:[2]
- Cirrosis.
- Retraso en el crecimiento y desarrollo del lenguaje.
- Retraso mental.
- Problemas motrices
- Dificultades para la orientación y percepción visual.
- En mujeres disfunción ovárica.
- Sepsis por E.Coli.
- Muerte si no se retira la galactosa de la dieta.
Tratamiento
El tratamiento consiste en:[3]
- Eliminar toda administración de la galactosa en la dieta. Esta se encuentra principalmente contenida en la leche.
- Se recomienda la administración de leche con proteínas a base de soja.
Referencias
- ↑ Marks’ Basic Medical Biochemistry. A Clinical Approach. Smith, C. et al. 2nd Edition. 2004. Lippincott Williams & Wilkins. ISBN 0-7817-2145-8.
- ↑ a b c d e f g Inborn Metabolic Diseases. Diagnosis and Treatment. Fernades, J. et al. 4th Edition. 2006. Springer. ISBN 978-3-540-28783-4.
- ↑ a b Diagnóstico y tratamiento de enfermedades metabólicas. Moreno, B. 1ª edición. 1997 Editorial Díaz de Santos S.A. ISBN 978-84-7978-303-7.
Véase también
Enlaces externos
Insights into the pathogenesis of galactosemia. Natural Course and Treatment of Neuropsychological Deficits in a Child with Early-treated Galactosemia. Parents of Galactosemic Children, Inc. Asociación española para la Galactosemia. A common mutation associated with the Duarte galactosemia allele. Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. ARHI: A new target of galactose toxicity in Classic Galactosemia.
Categorías:- Errores congénitos del metabolismo
- Trastornos nutricionales
- Tesaurismosis


Wikimedia foundation. 2010.