- Inhibidor enzimático
-
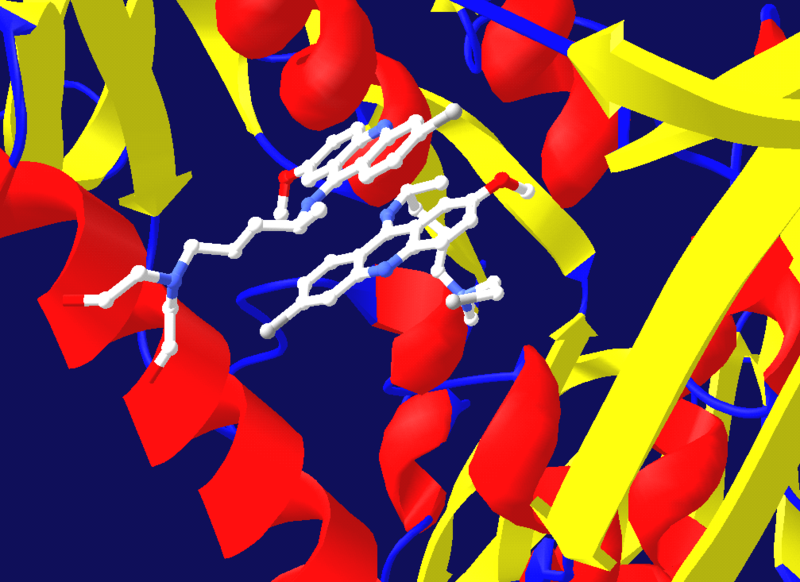
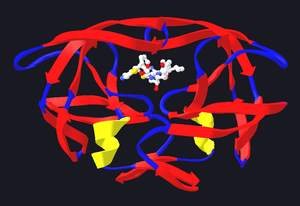 Modelo estructural de la proteasa del virus del SIDA unida a un inhibidor de la proteasa, el ritonavir. La estructura de la proteasa se muestra mediante cintas de color rojo, azul y amarillo, mientras que el inhibidor es representado por una estructura de esferas y varillas cerca del centro de la proteasa. Modelos creado a partir del PDB 1HXW.
Modelo estructural de la proteasa del virus del SIDA unida a un inhibidor de la proteasa, el ritonavir. La estructura de la proteasa se muestra mediante cintas de color rojo, azul y amarillo, mientras que el inhibidor es representado por una estructura de esferas y varillas cerca del centro de la proteasa. Modelos creado a partir del PDB 1HXW.
Los inhibidores enzimáticos son moléculas que se unen a enzimas y disminuyen su actividad. Puesto que el bloqueo de una enzima puede matar a un organismo patógeno o corregir un desequilibrio metabólico, muchos medicamentos actúan como inhibidores enzimáticos. También son usados como herbicidas y pesticidas. Sin embargo, no todas las moléculas que se unen a las enzimas son inhibidores; los activadores enzimáticos se unen a las enzimas e incrementan su actividad.
La unión de un inhibidor puede impedir la entrada del sustrato al sitio activo de la enzima y/u obstaculizar que la enzima catalice su reacción correspondiente. La unión del inhibidor puede ser reversible o irreversible. Normalmente, los inhibidores irreversibles reaccionan con la enzima de forma covalente y modifican su estructura química a nivel de residuos esenciales de los aminoácidos necesarios para la actividad enzimática. En cambio, los inhibidores reversibles se unen a la enzima de forma no covalente, dando lugar a diferentes tipos de inhibiciones, dependiendo de si el inhibidor se une a la enzima, al complejo enzima-sustrato o a ambos.
Muchos medicamentos son inhibidores enzimáticos, por lo que su descubrimiento y mejora es un campo de investigación activo en la bioquímica y la farmacología. La validez de un inhibidor enzimático medicinal suele venir determinada por su especificidad (su carencia de unirse a otras proteínas) y su potencia (su constante de disociación, la cual indica la concentración necesaria para inhibir a una enzima). Una alta especificidad y potencia asegura que el medicamento va a tener pocos efectos secundarios y por tanto una baja toxicidad.
Los inhibidores enzimáticos también son usados en la naturaleza y están implicados en la regulación del metabolismo. Por ejemplo, las enzimas en una ruta metabólica pueden ser inhibidas por los productos resultantes de sus respectivas rutas. Este tipo de retroalimentación negativa retarda el flujo a través de la ruta cuando los productos comienzan a acumularse y es una manera importante de mantener la homeostasis en una célula. Otros inhibidores enzimáticos celulares son proteínas que se unen específicamente e inhiben una diana enzimática. Esto puede ayudar a controlar enzimas que pueden ser dañinas para la célula, como las proteasas o nucleasas. Un buen ejemplo es el inhibidor de la ribonucleasa, que se une a esta enzima en una de las interacciones proteína–proteína más fuertes conocidas.[1] Como inhibidores enzimáticos naturales también cabe destacar los venenos, que son usados como defensa contra los depredadores o como forma de matar a una presa.
Contenido
Inhibidores ó sustractos reversibles
Los inhibidores reversibles se unen a las enzimas mediante interacciones no covalentes tales como los puentes de hidrógeno, las interacciones hidrofóbicas y los enlaces iónicos. Los enlaces débiles múltiples entre el inhibidor y el sitio activo se combinan para producir una unión fuerte y específica. Al contrario de lo que ocurre con el sustrato y los inhibidores irreversibles, los inhibidores reversibles generalmente no experimentan reacciones químicas cuando se unen a la enzima y pueden ser eliminados fácilmente por dilución o por diálisis.
Tipos de inhibidores reversibles
 Inhibición competitiva: el sustrato (S) y el inhibidor (I) compiten por el sitio activo.
Inhibición competitiva: el sustrato (S) y el inhibidor (I) compiten por el sitio activo.Existen tres tipos de inhibidores reversibles. Se clasifican en base al efecto producido por la variación de la concentración del sustrato de la enzima en el inhibidor.[2]
- En la inhibición competitiva, el sustrato y el inhibidor no se pueden unir a la misma enzima al mismo tiempo, como se muestra en la figura de la derecha. Esto generalmente ocurre cuando el inhibidor tiene afinidad por el sitio activo de una enzima en el que también se une el sustrato; el sustrato y el inhibidor compiten para el acceso al sitio activo de la enzima. Este tipo de inhibición se puede superar con concentraciones suficientemente altas del sustrato, es decir, dejando fuera de competición al inhibidor. Los inhibidores competitivos son a menudo similares en estructura al sustrato verdadero (ver ejemplos expuestos más abajo).
- En la inhibición mixta, el inhibidor se puede unir a la enzima al mismo tiempo que el sustrato. Sin embargo, la unión del inhibidor afecta la unión del sustrato, y viceversa. Este tipo de inhibición se puede reducir, pero no superar al aumentar las concentraciones del sustrato. Aunque es posible que los inhibidores de tipo mixto se unan en el sitio activo, este tipo de inhibición resulta generalmente de un efecto alostérico donde el inhibidor se une a otro sitio que no es el sitio activo de la enzima. La unión del inhibidor con el sitio alostérico cambia la conformación (es decir, la estructura terciaria o la forma tridimensional) de la enzima de modo que la afinidad del sustrato por el sitio activo se reduce.
- La inhibición no competitiva es una forma de inhibición mixta donde la unión del inhibidor con la enzima reduce su actividad pero no afecta la unión con el sustrato. Como resultado, el grado de inhibición depende solamente de la concentración de inhibidor.
Descripción cuantitativa de la inhibición reversible
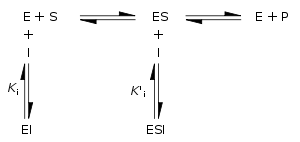
La inhibición reversible puede ser descrita cuantitativamente en términos de la unión del inhibidor a la enzima y al complejo enzima-sustrato, y sus efectos en las constantes cinéticas de la enzima. En el esquema clásico de Michaelis-Menten mostrado abajo, una enzima (E) se une a su sustrato (S) para formar el complejo enzima-sustrato ES. En la catálisis, este complejo se rompe para liberar el producto P y la enzima E. El inhibidor (I) puede unirse tanto a E como a ES con las constantes de disociación Ki o Ki', respectivamente.
- Los inhibidores competitivos se pueden unir a E, pero no a ES. La inhibición competitiva aumenta el valor de Km (es decir, el inhibidor interfiere con la unión del sustrato), pero no afecta a la Vmax (el inhibidor no obstaculiza la catálisis en ES porque no se puede unir a ES).
- Los inhibidores no competitivos tienen afinidades idénticas por E y ES (Ki = Ki'). La inhibición no competitiva no cambia la Km (es decir, no afecta a la unión del sustrato) pero disminuye la Vmax (es decir, la unión del inhibidor obstaculiza la catálisis).
- Los inhibidores de tipo mixto se unen tanto a E como a ES, pero sus afinidades por estas dos formas de enzimas son distintas (Ki ≠ Ki'). Por lo tanto, los inhibidores de tipo mixto interfieren con la unión del sustrato (incremento de Km) y dificulta la catálisis en el complejo ES (disminución de la Vmax).
 Esquema cinético aplicable a los inhibidores enzimáticos reversibles.
Esquema cinético aplicable a los inhibidores enzimáticos reversibles.Cuando una enzima tiene múltiples sustratos, los inhibidores pueden mostrar distintos tipos de inhibiciones dependiendo del sustrato que se considere, ya que el sitio activo posee dos diferentes lugares para la unión con el sustrato en el mismo sitio activo, uno para cada sustrato. Por ejemplo, un inhibidor puede competir con el sustrato A por el primer sitio de unión, pero ser un inhibidor no competitivo con respecto al sustrato B en el segundo sitio de unión.[3]
Medición de las constantes de disociación en un inhibidor reversible
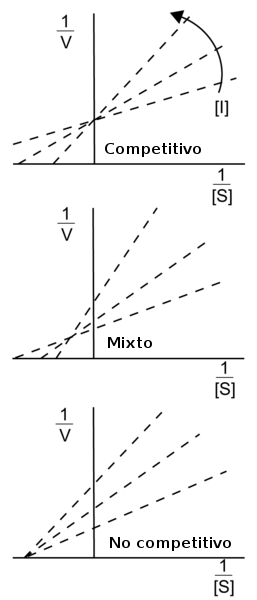
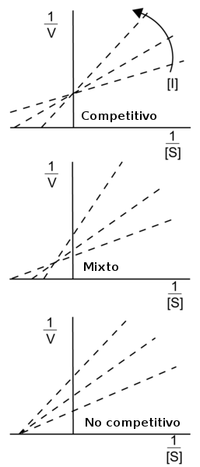 Diagramas de Lineweaver-Burke de los diferentes tipos de inhibidores enzimáticos reversibles. La flecha muestra el efecto producido por el incremento de las concentraciones de inhibidor.
Diagramas de Lineweaver-Burke de los diferentes tipos de inhibidores enzimáticos reversibles. La flecha muestra el efecto producido por el incremento de las concentraciones de inhibidor.Según lo observado arriba, un inhibidor enzimático está caracterizado por sus dos constantes de disociación, Ki y Ki', de la enzima y del complejo enzima-sustrato, respectivamente. La constante del complejo enzima-inhibidor Ki puede ser medida directamente por varios métodos. Un método extremadamente exacto es la calorimetría isoterma de titulación, en donde el inhibidor es titulado en una solución de enzimas y el calor liberado o absorbido es medido.[4] Sin embargo, la otra constante de disociación Ki' es difícil de medir directamente, ya que el complejo enzima-sustrato tiene un periodo de vida muy corto y está dando lugar a la reacción química para formar el producto. Por lo tanto, Ki' suele medirse de forma indirecta, observando la actividad enzimática bajo varias concentraciones de sustrato e inhibidor, y ajustando los datos[5] a una ecuación de Michaelis–Menten modificada:
donde los factores de modificación α y α' son definidos por la concentración del inhibidor y sus dos constantes de disociación
Así, en presencia del inhibidor, la efectividad de la enzima Km y Vmax es ahora (α/α')Km y (1/α')Vmax, respectivamente. Sin embargo, la ecuación de Michaelis-Menten modificada asume que la unión del inhibidor a la enzima alcanza el equilibrio, el cual puede ser un proceso muy lento para los inhibidores con constantes secundarias nanomolares de disociación. En estos casos, es usualmente más práctico tratar al inhibidor de unión fuerte como un inhibidor irreversible (ver abajo). Sin embargo, todavía puede ser posible estimar Ki' cinéticamente si Ki es medido independientemente.
Los efectos de diferentes tipos de inhibidores enzimáticos reversibles en la actividad enzimática pueden ser visualizados usando la representación gráfica de la ecuación de Michaelis–Menten, mediante los diagramas de Lineweaver-Burke o de Eadie-Hofstee. Por ejemplo, en los diagramas de Lineweaver-Burk a la derecha, las líneas de la inhibición competitiva intersecan el eje-y, ilustrando que tales inhibidores no afectan a la Vmax. De igual manera, las líneas de la inhibición no competitiva intersecan el eje-x, mostrando que estos inhibidores no afectan a la Km. Sin embargo, puede ser complicado estimar Ki y Ki' con precisión en estos diagramas,[6] por lo que es recomendable estimar estas constantes usando métodos más fiables de regresión no lineal, según lo descrito arriba.
Casos especiales
- El mecanismo de la inhibición parcialmente competitiva es similar al de la inhibición no competitiva, excepto que el complejo EIS tiene actividad catalítica, la cual decrece o incluso aumenta (activación parcialmente competitiva) en comparación al complejo enzima-sustrato (ES). Esta inhibición suele exhibir un valor más bajo de Vmax, pero un valor de Km inalterado.[7]
- La inhibición no competitiva se produce cuando el inhibidor se une sólo al complejo enzima-sustrato, no a la enzima libre. El complejo EIS es catalíticamente inactivo. Esta forma de inhibición es rara y causa una disminución tanto en el valor de Vmax como en el de Km.[7]
- La inhibición por sustrato y por producto es donde el sustrato o el producto de una reacción enzimática inhiben la actividad enzimática. Este tipo de inhibición puede seguir los patrones competitivos, no competitivos o mixtos. En la inhibición por sustrato hay una disminución progresiva de la actividad a altas concentraciones de sustrato. Esto puede indicar la existencia de dos sitios de unión entre sustrato y enzima. Cuando hay poco sustrato, se ocupa el sitio de alta afinidad y sigue la cinética normal. Sin embargo, a altas concentraciones, el segundo sitio de inhibición se ocupa, inhibiendo a la enzima.[8] La inhibición por parte del producto es a menudo una característica reguladora en el metabolismo y puede ser una forma de retroalimentación negativa.
- La inhibición lenta y fuerte se produce cuando el complejo enzima-inhibidor EI inicial experimenta una isomerización a un segundo complejo más fuertemente unido, EI*, pero el proceso total de la inhibición es reversible. Esto se manifiesta como un lento aumento en la inhibición enzimática. En estas condiciones, la tradicional cinética de Michaelis–Menten da un valor falso para Ki, el cual depende del tiempo. El verdadero valor de Ki puede ser obtenido a través de un análisis más complejo de las constantes de rango de encendido (kon) y apagado (koff) para la asociación del inhibidor (véase la inhibición irreversible para más información).
Ejemplos de inhibidores reversibles
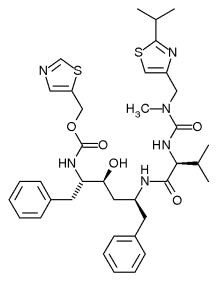
Puesto que las enzimas han evolucionado para unirse a sus sustratos fuertemente, y la mayoría de los inhibidores reversibles se unen al sitio activo de las enzimas, es poco sorprendente que algunos de estos inhibidores sean muy similares en estructura a los sustratos de sus dianas. Como ejemplo de estos imitadores de sustratos caben destacar los inhibidores de la proteasa, una clase muy efectiva de fármacos antirretrovirales usados para tratar el VIH.[9] La estructura del ritonavir (figura de la derecha), un inhibidor de la proteasa, consiste en un péptido con tres enlaces peptídicos. Dicha estructura se asemeja a la proteína que es el sustrato de la proteasa del VIH, por lo que ambos compiten por la unión al sitio activo de la enzima.
Los inhibidores enzimáticos son a menudo diseñados para imitar el estado de transición o intermedio de una reacción catalizada por una enzima. Esto asegura que el inhibidor cambie el estado de transición estableciendo un efecto en la enzima, lo que resulta en una afinidad de unión mejor (baja Ki) que los diseños basados en sustratos. Un ejemplo de un inhibidor en ese estado de transición es el fármaco antiviral oseltamivir, que imita la naturaleza plana del anillo del ion oxonio en la reacción de la neuraminidasa, una enzima del virus.[10]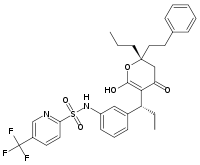 Estructura molecular del tipranavir, un inhibidor de proteasa de carácter no peptídico.
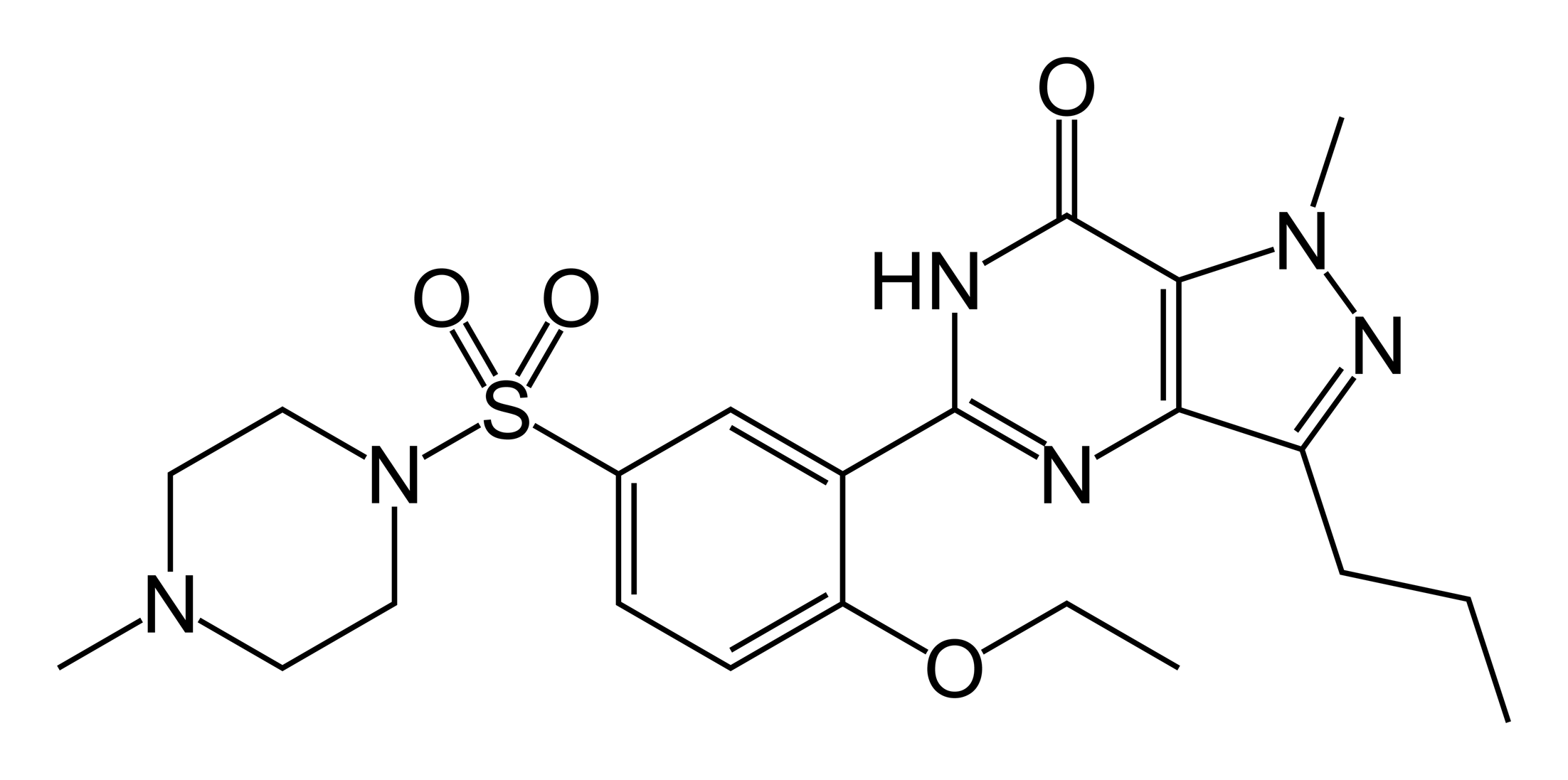
Estructura molecular del tipranavir, un inhibidor de proteasa de carácter no peptídico.Sin embargo, no todos los inhibidores están basados en la estructura del sustrato. Por ejemplo, la estructura de otro inhibidor de la proteasa del VIH, el tipranavir, (representada a la derecha), no está basada en un péptido y no tiene similitudes estructurales obvias con la proteína sustrato. Estos inhibidores no peptídicos pueden ser más estables que los inhibidores que contienen enlaces peptídicos porque estos no son sustratos para las peptidasas, con lo que son menos propensas a ser degradadas en la célula.[11]
En el diseño de fármacos es importante considerar las concentraciones de sustrato a las cuales se expondrá la enzima en cuestión. Por ejemplo, algunos inhibidores de proteínas quinasas tienen estructuras químicas que son similares al adenosín trifosfato, uno de los sustratos de esta enzima. Sin embargo, ciertos fármacos que son simplemente inhibidores competitivos tendrán que competir con altas concentraciones de ATP en la célula. Las proteínas quinasas también pueden ser inhibidas por competencia en el sitio de unión donde la quinasa interactúa con sus proteínas sustrato, y la mayoría de las proteínas presentes en el interior de una célula se encuentran a concentraciones mucho menores que las concentraciones de ATP. En consecuencia, si dos inhibidores de proteínas quinasas se unen en sus sitios activos con afinidad similar, pero solo uno tiene que competir con el ATP, entonces el inhibidor competitivo en el sitio de unión de la proteína inhibirá a la enzima más eficientemente.[12]
Inhibidores irreversibles
 Reacción del inhibidor irreversible diisopropilfluorofosfato (DFP) con una serín-proteasa.
Reacción del inhibidor irreversible diisopropilfluorofosfato (DFP) con una serín-proteasa.Los inhibidores irreversibles normalmente modifican una enzima covalentemente, con lo que la inhibición no puede ser invertida. Los inhibidores irreversibles suelen contener grupos funcionales reactivos como mostazas nitrogenadas, aldehídos, haloalcanos o alquenos. Estos grupos electrofílicos reaccionan con las cadenas de aminoácidos para formar uniones covalentes. Los residuos modificados son aquellos que contienen en sus cadenas laterales nucleófilos como por ejemplo un grupo hidroxilo o un grupo sulfhidrilo. Esto incluye a los aminoácidos serina (como en el DFP, a la derecha), cisteína, treonina o tirosina.[13]
Tipos de inhibiciones irreversibles
La inhibición irreversible es diferente de la inactivación enzimática reversible. Los inhibidores irreversibles son generalmente específicos para un tipo de enzima y no inactivan a todas las proteínas. No funcionan destruyendo la estructura proteínica, sino alterando específicamente la estructura tridimencional del sitio activo inhabilitándolo. Por ejemplo, el pH y las temperaturas extremas causan la desnaturalización de casi todas las proteínas, pero este no es un efecto específico. De forma similar, algunos tratamientos químicos no específicos destruyen la estructura de la proteína: por ejemplo, si son sometidas a una elevada concentración de ácido clorhídrico, el cual hidrolizará los enlaces peptídicos que mantienen unidos los aminoácidos de las proteínas.[14]
Los inhibidores irreversibles dan lugar a una inhibición dependiente del tiempo y, por ello, su potencia no puede ser caracterizada mediante la determinación del valor editar] Análisis de la inhibición irreversible
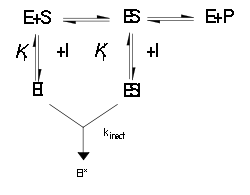 Esquema de la cinética enzimática de los inhibidores irreversibles.
Esquema de la cinética enzimática de los inhibidores irreversibles.Como se muestra en la figura de la izquierda, los inhibidores irreversibles forman inicialmente un complejo reversible y no covalente con la enzima (EI o ESI), que reaccionará posteriormente para producir una modificación covalente en lo que se denomina el "complejo del punto muerto" EI*. La tasa a la cual se forma EI* es llamada tasa de inactivación o kinact. Puesto que la formación de EI puede competir con ES, la unión de los inhibidores irreversibles puede ser prevenida por competencia tanto con el sustrato como con un segundo inhibidor reversible. Este efecto de protección es una buena evidencia de una reacción específica del inhibidor irreversible con el sitio activo.
Los pasos de unión e inactivación de esta reacción son analizados incubando la enzima con el inhibidor y midiendo la actividad que va quedando a lo largo del tiempo. La actividad irá disminuyendo de forma paulatina con tiempo, generalmente siguiendo una dinámica de decaimiento exponencial. Ajustando estos datos a una ecuación de rango podemos obtener la tasa de inactivación a esta concentración de inhibidor. Esto se hace a diferentes concentraciones de inhibidor. Si un complejo EI reversible está involucrado el rango de inactivación será saturable y al ajustarla a esta curva obtendremos los valores de kinact y Ki.[16]
Otro método que es ampliamente utilizado en estos análisis es la espectrometría de masas. En este caso, la medida exacta de la masa de la enzima nativa sin modificar y de la enzima inactivada, nos da el aumento en la masa causado por la reacción con el inhibidor, con lo que obtenemos la estequiometría de la reacción.[17] Para llevar a cabo esta técnica suele ser necesario el uso de un espectrómetro de masas MALDI-TOF. Una técnica complementaria a esta es la huella peptídica, que implica la digestión de proteínas nativas o modificadas con una peptidasa como la tripsina. Esto genera una serie de péptidos que pueden ser analizados usando un espectrómetro de masas. El péptido que cambie su masa después de llevar a cabo la reacción con el inhibidor será aquel que contenga el sitio de la modificación.
Casos especiales
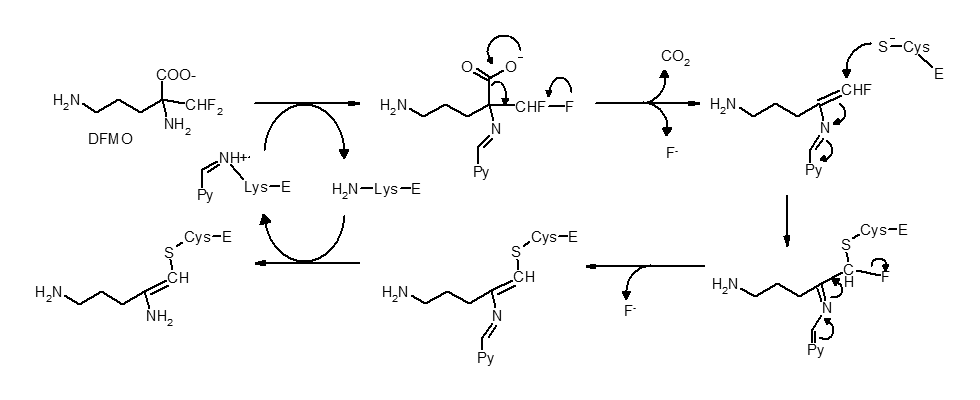
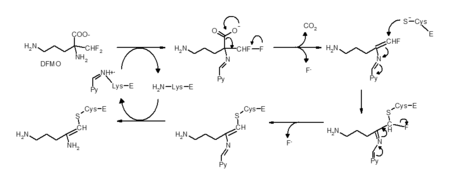 Mecanismo químico para inhibición irreversible de la ornitina descarboxilasa por medio de DFMO. El piridoxal 5'-fosfato (Py) y la enzima (E) no se muestran.[18] (Adaptado del artículo que figura en la referencia).
Mecanismo químico para inhibición irreversible de la ornitina descarboxilasa por medio de DFMO. El piridoxal 5'-fosfato (Py) y la enzima (E) no se muestran.[18] (Adaptado del artículo que figura en la referencia).No todos los inhibidores irreversibles forman uniones covalentes con la enzima que tienen como diana. Algunos inhibidores reversibles se unen tan fuertemente a su objetivo que son prácticamente irreversibles. Estos inhibidores de unión fuerte suelen mostrar una cinética similar a la de los inhibidores irreversibles que forman enlaces covalentes. En estos casos, algunos de estos inhibidores se unen rápidamente a la enzima formando un complejo EI de baja afinidad, que después experimenta una reacción más lenta hacia un complejo EI* muy fuertemente unido (ver la imagen arriba). Este comportamiento cinético es denominado unión lenta.[19] Este lento reajuste posterior normalmente implica un cambio conformacional cuando la enzima se pliega alrededor de la molécula inhibidora. Como ejemplos de inhibidores de unión lenta cabe destacar algún fármaco importante como el metotrexato,[20] el alopurinol[21] y la forma activada del aciclovir.[22]
Ejemplos de inhibidores irreversibles
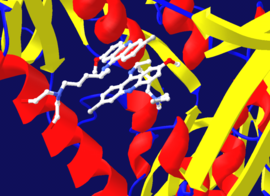 Tripanotión reductasa donde se muestran dos moléculas unidas al centro activo: la inferior es un inhibidor unido de forma irreversible y la superior un inhibidor unido de forma reversible. Creado mediante PDB 1GXF.
Tripanotión reductasa donde se muestran dos moléculas unidas al centro activo: la inferior es un inhibidor unido de forma irreversible y la superior un inhibidor unido de forma reversible. Creado mediante PDB 1GXF.El diisopropilfluorofosfato (DFP) se muestra como un ejemplo de inhibidor irreversible de la proteasa en el apartado "Inhibidores irreversibles" arriba a la derecha. La enzima hidroliza el enlace entre el fósforo y el flúor, pero el residuo de fosfato se mantiene unido a una serina en el centro activo, inactivándolo.[23] Además, el DFP también reacciona con el centro activo de la acetilcolinesterasa en la sinapsis de las neuronas, lo que lo convierte en una potente neurotoxina, con una dosis letal a partir de cantidades inferiores a 100 mg.[24]
La inhibición suicida es un tipo común de inhibición irreversible donde la enzima convierte al inhibidor en una sustancia reactiva en su centro activo. Un ejemplo de esto es el inhibidor de biosintetizadores de poliaminas α-difluorometilornitina o DFMO, que es un análogo del aminoácido ornitina, y es usado para tratar la Tripanosomiasis Africana (enfermedad del sueño). La ornitina decarboxilasa puede catalizar la descarboxilación del DFMO sustituyendo a la ornitina, como se muestra en la figura del apartado anterior. Sin embargo, esta reacción de descarboxilación es seguida por la eliminación del átomo de flúor, lo que convierte a este intermediario en una imina, una especie altamente electrofílica. Esta forma reactiva del DFMO reacciona posteriormente con un residuo de cisteína o lisina en el centro activo para inactivar la enzima irreversiblemente.[18]
Puesto que la inhibición irreversible implica a menudo la formación inicial de un complejo EI no covalente, a veces es posible que un inhibidor pueda unirse a una enzima de diversas formas. Como ejemplo cabe destacar el caso de la enzima tripanotión reductasa perteneciente al protozoo y parásito humano Trypanosoma cruzi, donde dos moléculas de un inhibidor llamado mostaza quinacrina, presentan la capacidad de unirse a su centro activo. La molécula superior se une de forma reversible, pero la de abajo se une de forma covalente al reaccionar con un residuo de aminoácido a través de su grupo de mostaza nitrogenada.[25]
Descubrimiento y diseño de inhibidores
La investigación y desarrollo de nuevos fármacos es un largo proceso en el cual el primer paso casi siempre es el descubrimiento de un nuevo inhibidor enzimático. En el pasado, la única forma de descubrir estos nuevos inhibidores era mediante el sistema de prueba y error: probando un elevado número de compuestos contra la enzima a la cual se quiere inhibir y esperando conseguir alguno que sea efectivo. Esta aproximación, si bien se basa en la fuerza bruta, sigue logrando actualmente un gran éxito gracias a nuevos sistemas de barrido, como la química combinatoria, que rápidamente producen un gran número de compuestos novedosos, y a la tecnología basada en la investigación de procesamiento de alto-rendimiento, mediante la cual se pueden revisar rápidamente estas enormes bibliotecas químicas de inhibidores útiles.[26]
Más recientemente, se ha comenzado a aplicar un nuevo tipo de investigación alternativa: el diseño racional de fármacos que usa la estructura tridimensional del sitio activo de una enzima para predecir qué moléculas podrían ser inhibidores.[27] Una vez realizada la predicción, se prueban y se selecciona el mejor. Este nuevo inhibidor es usado después para tratar de obtener una estructura de la enzima en un complejo enzima-sutrato para mostrar cómo se une la molécula al sitio activo. Esta estructura es después inspeccionada y se llevan a cabo ciertos cambios en la estructura del inhibidor con el fin de optimizar la unión con la enzima. Este ciclo de prueba y optimización se repite de forma sucesiva hasta obtener un inhibidor lo suficientemente potente, es decir, cuando se alcanza un valor de la constante de disociación que esté en torno a <10-9 M.[28]
Aplicaciones de los inhibidores
Los inhibidores enzimáticos pueden ser encontrados en la naturaleza, pero también son diseñados y producidos como parte de la farmacología y la bioquímica. Los venenos naturales son a menudo inhibidores enzimáticos que han evolucionado para defender a una planta o animal contra sus depredadores. Estas toxinas naturales incluyen algunos de los compuestos más venenosos conocidos hasta hoy. Los inhibidores artificiales son a menudo usados como medicamentos, pero también existen algunos utilizados como insecticidas (como el malathion), herbicidas (como el glifosato) o desinfectantes, (como el triclosán).
Quimioterapia
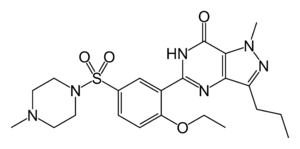 Estructura del sildenafil (Viagra).
Estructura del sildenafil (Viagra).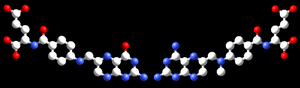 Comparación de la estructura del ácido fólico, un coenzima (izquierda), con la estructura del metotrexato, un fármaco anticancerígeno (derecha).
Comparación de la estructura del ácido fólico, un coenzima (izquierda), con la estructura del metotrexato, un fármaco anticancerígeno (derecha).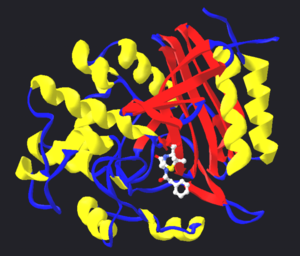 Estructura tridimensional del complejo formado por la penicilina G unida a la transpeptidasa de Streptomyces. Generado mediante PDB 1PWC.
Estructura tridimensional del complejo formado por la penicilina G unida a la transpeptidasa de Streptomyces. Generado mediante PDB 1PWC.Los inhibidores enzimáticos son utilizados principalmente como fármacos en el tratamiento de diversas enfermedades. Muchos de estos inhibidores son capaces de actuar sobre enzimas humanas y así corregir determinadas patologías. Sin embargo, no todos los fármacos son inhibidores enzimáticos. Algunos de ellos, tales como los fármacos anti-epilépticos, alteran la actividad enzimática de forma indirecta, aumentando o disminuyendo la síntesis de dicha enzima. Estos efectos son denominados inducción e inhibición enzimática y consisten en alteraciones en el patrón de expresión génica, lo cual no está relacionado con el tipo de inhibición enzimática discutido aquí. Otras drogas interactúan con otras dianas celulares que no son enzimas, como los canales iónicos o los receptores de membrana.
Un ejemplo de inhibidor enzimático terapéutico es el sildenafil (Viagra), utilizado como tratamiento de la disfunción eréctil. Este compuesto es un potente inhibidor de la fosfodiesterasa tipo 5 específica de GMPc, una enzima que degrada una molécula de señalización celular, el GMPc.[29] Esta molécula de señalización es la responsable de la relajación del músculo liso y permite el flujo de sangre hacia el interior de los cuerpos cavernosos del pene, lo cual causa la erección. El sildenafil inhibe la actividad de la enzima que degrada la señal, el GMPc, uniéndose al mismo sitio que éste debido a su similaridad estructural. Esto permite que el GMPc no sea degradado y pueda permanecer activo durante períodos más largos de tiempo.
Otro ejemplo de similaridad estructural entre inhibidores y sustratos enzimáticos es que el que se muestra en la figura de la derecha, entre el metotrexato, un fármaco, y el ácido fólico, un coenzima. El ácido fólico es la forma oxidada del sustrato de la dihidrofolato reductasa, una enzima implicada en la biosíntesis de timidina, purinas y aminoácidos. El metotrexato es un potente inhibidor de esta enzima que, al estar relacionada con la síntesis de nucleótidos, presenta una toxicidad específica de aquellas células con una rápida tasa de crecimiento. Por ello, el metotrexato se utiliza a menudo como fármaco anticancerígeno en quimioterapia.[30]
Otro tipo de inhibidores enzimáticos son utilizados con el fin de inhibir aquellas enzimas necesarias para la supervivencia de patógenos. Por ejemplo, las bacterias presentan una gruesa pared celular compuesta principalmente de un polímero denominado peptidoglicano. Ciertos antibióticos como la penicilina y la vancomicina inhiben a la enzima responsable de la producción y el entrecruzamiento de las hebras de peptidoglicano,[31] lo cual da lugar a una pérdida de fuerza de la pared celular y, por consiguiente, a la lisis de la célula, incapaz ahora de resistir la elevada presión osmótica. En la figura se puede apreciar una molécula de penicilina unida a su diana, la transpeptidasa de la bacteria Streptomyces R61 (la proteína se muestra en un formato de cintas y la penicilina en un formato de esferas y barras). También se utilizan otro tipo de toxicidades selectivas mediante antibióticos que aprovechan las diferencias presentes en la estructura de los ribosomas o en la síntesis de ácidos grasos.
El diseño de fármacos se ve muy facilitado en estos casos en los que la enzima diana es esencial para la supervivencia del patógeno y no está presente o es muy diferente en humanos.
Control metabólico
Los inhibidores enzimáticos son también importantes a nivel del control metabólico. Muchas de las rutas metabólicas que tienen lugar en la célula son inhibidas por metabolitos que controlan la actividad enzimática mediante procesos de regulación alostérica o inhibición por sustrato. A modo de ejemplo cabe destacar la regulación alostérica de la glucólisis. Esta ruta catbólica consume glucosa y produce ATP, NADH y piruvato. Una de las etapas clave en la regulación de la glucolisis es la reacción catalizada por la fosfofructoquinasa-1 (PFK1). Cuando los niveles de ATP aumentan, el ATP se une a un sitio alostérico de la PFK1, con lo que se reduce la actividad de la enzima, la glucolisis se inhibe y los niveles de ATP se reducen de nuevo. Este control por retroalimentación negativa (feedback negativo) ayuda a mantener los niveles de ATP constantes en la célula. Sin embargo, las rutas metabólicas no están únicamente reguladas por medio de la inhibición, la activación enzimática es igualmente importante. La enzima PFK1 presenta activadores alostéricos, como son la fructosa 2,6-bifosfato y el ADP.[32]
La inhibición enzimática fisiológica también puede ser producida por inhibidores proteicos específicos. Este mecanismo se puede observar en el páncreas, donde se sintetizan multitud de precursores de enzimas digestivas denominadas zimógenos. Muchos de ellos son activados por la tripsina, una proteasa, por lo que es muy importante inhibir la actividad de la tripsina en el páncreas con el fin de prevenir fenómenos de autodigestión. Uno de los mecanismos para mantener la tripsina inactiva es la producción de un potente y específico inhibidor de tripsina en el páncreas. Este inhibidor se une con una alta afinidad a la tripsina, previniendo así su actividad.[33] Aunque el inhibidor de tripsina es una proteína, no es hidrolizado por la propia tripsina, ya que elimina las moléculas de agua de su centro activo y desestabiliza el estado de transición.[34] Otros ejemplos de inhibidores enzimáticos fisiológicos son el inhibidor barstar, cuya función es inhibir la actividad de una ribonucleasa bacteriana denominada barnasa,[35] y los inhibidores de las protein-fosfatasas.[36]
Inhibidores de la acetilcolinesterasa
 Con el fin de disuadir a los depredadores de semillas, las legumbres incorporan inhibidores de tripsina, que interfieren con la digestión.
Con el fin de disuadir a los depredadores de semillas, las legumbres incorporan inhibidores de tripsina, que interfieren con la digestión.La acetilcolinesterasa (AChE) es una enzima que se encuentra en los animales, desde los insectos hasta los humanos. Es esencial en la actividad que desempeñan las neuronas, ya que su función consiste en la ruptura del neurotransmisor acetilcolina en sus constituyentes, acetato y colina. Es una caso único entre los neurotransmisores, ya que la mayoría de ellos, como la serotonina, la dopamina o la noradrenalina, son reabsorbidos en la brecha sináptica. Existe un amplio número de inhibidores de la AChE que son utilizados tanto en el campo de la medicina como en el campo de la agricultura. Algunos de estos inhibidores son reversibles, como el edrofonio, la fisostigmina, y la neostigmina, los cuales son utilizados en el tratamiento de la miastenia gravis y en la aplicación de anestesia. Los pesticidas del tipo carbamato son otro ejemplo de inhibidores reversibles de la AChE. Como ejemplo de inhibidores irreversibles de la AChE caben destacar los insecticidas organofosforados, como el malathion, el paratión y los clorpirifós.
Venenos naturales
Tanto algunos animales como algunas plantas son capaces de sintetizar una amplia gama de sustancias venenosas de diverso origen: metabolitos secundarios, péptidos y proteínas, que pueden actuar como inhibidores enzimáticos. Las toxinas naturales suelen ser pequeñas moléculas orgánicas con tanta diversidad que probablemente existan inhibidores para la mayoría de los procesos metabólicos.[37] Dicha inhibición puede producirse a diferentes niveles en la célula: inhibición de receptores de membrana, de canales iónicos o de proteínas estructurales. Por ejemplo, el paclitaxel (taxol), una molécula orgánica obtenida del tejo (Taxus), se une con una elevada afinidad a los dímeros de tubulina, impidiendo así el proceso de polimerización de los microtúbulos, crucial, entre otras cosas, en la división celular.[38]
Muchos de estos venenos naturales actúan como neurotoxinas capaces de causar parálisis que pueden llevar a la muerte, pudiendo ser utilizados como estrategia defensiva frente a los depredadores o como sistema de caza en la captura de presas. Algunos de estos inhibidores naturales, a pesar de sus características tóxicas, son muy valorados por sus potenciales usos terapéuticos cuando son administrados en dosis adecuadas.[39] Como ejemplo de neurotoxina cabe destacar los glicoalcaloides, obtenidos a partir de las plantas pertenecientes a la familia Solanaceae (entre las que se incluyen la patata, el tomate y la berenjena), que son inhibidores de la acetilcolinesterasa. La inhibición de esta enzima causa un aumento descontrolado de los niveles del neurotransmisor acetilcolina, lo que conlleva parálisis muscular y muerte. La neurotoxicidad también puede ser el resultado de la inhibición de receptores, como en el caso de la atropina, obtenida a partir de la especie Atropa belladonna, que funciona como un antagonista competitivo de los receptores muscarínicos de acetilcolina.[40]
Aunque muchas de las toxinas naturales son metabolitos secundarios, algunas son péptidos o proteínas. Como ejemplo cabe destacar a la toxina peptídica alfa-amanitina, encontrada en los hongos de la especie Amanita phalloides, que es un potente inhibidor enzimático capaz de impedir la actividad de la ARN polimerasa II en la transcripción del ADN.[41] Otra toxina peptídica es la microcistina, encontrada en ciertas algas y capaz de inhibir ciertas protein-fosfatasas.[42] Esta toxina puede contaminar las reservas de agua si las algas que la producen alcanzan determinados niveles de concentración. Se ha demostrado su alto potencial carcinogénico y su capacidad de causar hemorragia hepática aguda y muerte tras una exposición a altas dosis.[43]
Las proteínas también pueden llegar a ser venenos naturales, como es el caso del inhibidor de tripsina (discutido anteriormente) encontrado en algunas legumbres. Menos comunes son las enzimas tóxicas. Este tipo de enzimas actúan como inhibidores irreversibles de dianas enzimáticas que modifican químicamente sus sustratos enzimáticos. Un ejemplo es el ricino, una proteína tóxica extremadamente potente, que se encuentra en la planta Ricinus communis. Esta enzima es una glicosidasa que inactiva a los ribosomas. Como el ricino es un inhibidor catalítico irreversible, esto permite que tan solo una molécula de ricino pueda matar una célula.[44]
Véase también
Notas
- ↑ Shapiro R, Vallee BL. Interaction of human placental ribonuclease with placental ribonuclease inhibitor. Biochemistry. 1991 Feb 26;30(8):2246–55. PMID 1998683
- ↑ Berg J., Tymoczko J. and Stryer L. (2002) Biochemistry. W. H. Freeman and Company ISBN 0-7167-4955-6
- ↑ *Irwin H. Segel, Enzyme Kinetics : Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley–Interscience; New edition (1993), ISBN 0-471-30309-7
- ↑ Holdgate GA. Making cool drugs hot: isothermal titration calorimetry as a tool to study binding energetics. Biotechniques. 2001 Jul;31(1):164–6 PMID 11464510
- ↑ Leatherbarrow RJ. Using linear and non-linear regression to fit biochemical data. Trends Biochem Sci. 1990 Dec;15(12):455–8. PMID 2077683
- ↑ Tseng SJ, Hsu JP. A comparison of the parameter estimating procedures for the Michaelis–Menten model. J Theor Biol. 1990 Aug 23;145(4):457–64. PMID 2246896
- ↑ a b Irwin H. Segel, Enzyme Kinetics : Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley-Interscience; New Ed edition (1993), ISBN 0-471-30309-7
- ↑ Dixon, M. Webb, E.C., Thorne, C.J.R. and Tipton K.F., Enzymes (3rd edition) Longman, London (1979) See p. 126
- ↑ Hsu JT, Wang HC, Chen GW, Shih SR. Antiviral drug discovery targeting to viral proteases. Curr Pharm Des. 2006; 12(11):1301–14. PMID 16611117
- ↑ Lew W, Chen X, Kim CU (2000). «Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor». Curr. Med. Chem. 7 (6): pp. 663–72. PMID 10702632.
- ↑ Fischer PM (2003). «The design, synthesis and application of stereochemical and directional peptide isomers: a critical review». Curr. Protein Pept. Sci. 4 (5): pp. 339–56. PMID 14529528.
- ↑ Bogoyevitch MA, Barr RK, Ketterman AJ. Peptide inhibitors of protein kinases—discovery, characterisation and use. Biochim Biophys Acta. 2005 Dec 30;1754(1-2):79–99. PMID 16182621
- ↑ Lundblad R. L. Chemical Reagents for Protein Modification CRC Press Inc (2004) ISBN 0-8493-1983-8
- ↑ N. Price, B. Hames, D. Rickwood (Ed.) Proteins LabFax Academic Press (1996) ISBN 0-12-564710-7
- ↑ Adam GC, Cravatt BF, Sorensen EJ. (2001) Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem. Biol. 8(1):81-95.
- ↑ Maurer T, Fung HL. Comparison of Methods for Analyzing Kinetic Data From Mechanism-Based Enzyme Inactivation: Application to Nitric Oxide Synthase. AAPS PharmSci. (2000) 2(1)E8. PMID 11741224
- ↑ Loo JA, DeJohn DE, Du P, Stevenson TI, Ogorzalek Loo RR (1999). «Application of mass spectrometry for target identification and characterization». Med Res Rev 19 (4): pp. 307–19. PMID 10398927.
- ↑ a b Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. J Biol Chem. 1992 Jan 5;267(1):150–8. PMID 1730582
- ↑ Szedlacsek, S.E. and Duggleby, R.G. Kinetics of slow and tight-binding inhibitors. Meth. Enzymol., (1995) 249: 144–180. PMID 7791610
- ↑ Stone SR, Morrison JF. Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues. Biochim Biophys Acta. 1986 Feb 14;869(3):275–85. PMID 3511964
- ↑ Hille R, Massey V. Tight binding inhibitors of xanthine oxidase. Pharmacol Ther. 1981;14(2):249–63. PMID 4322209
- ↑ Reardon JE. Herpes simplex virus type 1 and human DNA polymerase interactions with 2'-deoxyguanosine 5'-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J Biol Chem. 1989 Nov 15;264(32):19039–44. PMID 2553730
- ↑ J. A. Cohen , R. A. Oosterbaan and F. Berends Organophosphorus compounds Meth. Enzymol. (1967) 11, 686
- ↑ Brenner, G. M. (2000): Pharmacology. Philadelphia, PA: W.B. Saunders Company. ISBN 0-7216-7757-6
- ↑ Saravanamuthu A, Vickers TJ, Bond CS, Peterson MR, Hunter WN, Fairlamb AH. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: a template for drug design. J Biol Chem. 2004 Jul 9;279(28):29493–500. PMID 15102853
- ↑ Koppitz M, Eis K (2006). «Automated medicinal chemistry». Drug Discov. Today 11 (11-12): pp. 561–8. doi:. PMID 16713909.
- ↑ Scapin G (2006). «Structural biology and drug discovery». Curr. Pharm. Des. 12 (17): pp. 2087–97. doi:. PMID 16796557.
- ↑ Hunter WN. Rational drug design: a multidisciplinary approach. Mol Med Today. 1995 Apr;1(1):31, 34. PMID 9415135
- ↑ Maggi M, Filippi S, Ledda F, Magini A, Forti G. Erectile dysfunction: from biochemical pharmacology to advances in medical therapy. Eur J Endocrinol. 2000 Aug;143(2):143–54 PMID 10913932
- ↑ McGuire JJ. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9(31):2593–613. PMID 14529544
- ↑ Katz AH, Caufield CE. Structure-based design approaches to cell wall biosynthesis inhibitors. Curr Pharm Des. 2003;9(11):857–66. PMID 12678870
- ↑ Okar DA, Lange AJ. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors. 1999;10(1):1–14.
- ↑ Nicholas Price, Lewis Stevens, Fundamentals of Enzymology, Oxford University Press, (1999) ISBN 0-19-850229-X
- ↑ Smyth TP. Substrate variants versus transition state analogues as noncovalent reversible enzyme inhibitors. Bioorg Med Chem. 2004 Aug 1;12(15):4081–8. PMID 15246086
- ↑ Hartley RW. Barnase and barstar: two small proteins to fold and fit together. Trends Biochem Sci. 1989 Nov;14(11):450–4. PMID 2696173
- ↑ Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998 Sep 1;3:D961–72. PMID 9727084
- ↑ Tan G, Gyllenhaal C, Soejarto DD. Biodiversity as a source of anticancer drugs. Curr Drug Targets. 2006 Mar;7(3):265-77. PMID 16515527
- ↑ Abal M, Andreu JM, Barasoain I. Taxanes: microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr Cancer Drug Targets. 2003 Jun;3(3):193–203. PMID 12769688
- ↑ Hostettmann K, Borloz A, Urbain A, Marston A, Natural Product Inhibitors of Acetylcholinesterase Current Organic Chemistry, 2006 May;10(8):825–47
- ↑ DeFrates LJ, Hoehns JD, Sakornbut EL, Glascock DG, Tew AR. Antimuscarinic intoxication resulting from the ingestion of moonflower seeds. Ann Pharmacother. 2005 Jan;39(1):173-6. PMID 15572604
- ↑ Vetter J. Toxins of Amanita phalloides. Toxicon. 1998 Jan;36(1):13–24. PMID 9604278
- ↑ Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN. Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem. 2002 Nov;9(22):1981–9. PMID 12369866
- ↑ Bischoff K. The toxicology of microcystin-LR: occurrence, toxicokinetics, toxicodynamics, diagnosis and treatment. Vet Hum Toxicol. 2001 Oct;43(5):294-7. PMID 11577938
- ↑ Hartley MR, Lord JM. Cytotoxic ribosome-inactivating lectins from plants. Biochim Biophys Acta. 2004 Sep 1;1701(1–2):1–14. PMID 15450171
Enlaces externos
- Tutorial sobre inhibición enzimática
- Simbología y terminología en la cinética enzimática
- Base de datos de fármacos e inhibidores enzimáticos (PubChem del NCBI
- BRENDA: base de datos de enzimas y sus correspondientes inhibidores conocidos
- Enzimas, Cinética y Diagnóstico. Aplicaciones médicas de los inhibidores enzimáticos
- BindingDB: base de datos pública que recoge los valores de afinidades de unión proteína-ligando
- Ejercicios animados de inhibición enzimática: tutoriales y preguntas



![V = \frac{V_{max}[S]}{\alpha K_{m} + \alpha^{\prime}[S]} = \frac{(1/\alpha^{\prime})V_{max}[S]}{(\alpha/\alpha^{\prime}) K_{m} + [S]}](6/0561ea39d79a20421e46a45ccc345d61.png)
![\alpha = 1 + \frac{[I]}{K_{i}}](0/280e85eac3f83f2370a658fe52217471.png)
![\alpha^{\prime} = 1 + \frac{[I]}{K_{i}^{\prime}}](7/857b64382bd94382e988210bb7a6d772.png)




Wikimedia foundation. 2010.