- Alineamiento estructural
-
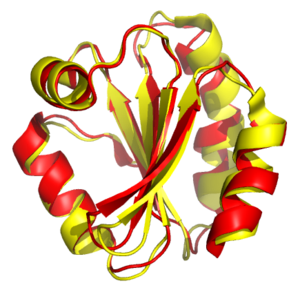 Alineamiento estructural de tiorredoxinas del ser humano y de la mosca Drosophila melanogaster. Las proteínas se muestran como cintas, con la proteína humana en rojo y la de la mosca en amarillo. Generado con PDB 3TRX y 1XWC.
Alineamiento estructural de tiorredoxinas del ser humano y de la mosca Drosophila melanogaster. Las proteínas se muestran como cintas, con la proteína humana en rojo y la de la mosca en amarillo. Generado con PDB 3TRX y 1XWC.
Un alineamiento estructural es un tipo de alineamiento de secuencias basado en la comparación de la forma. Estos alineamientos intentan establecer equivalencias entre dos o más estructuras de polímeros basándose en su forma y conformación tridimensional. El proceso se aplica normalmente a las estructuras terciarias de las proteínas, pero también puede usarse para largas moléculas de ARN. En contraste a la simple superposición estructural, donde al menos se conocen algunos residuos equivalentes de las dos estructuras, el alineamiento estructural no requiere un conocimiento previo de posiciones equivalentes. Es una valiosa herramienta para la comparación de proteínas con baja similitud entre sus secuencias, en donde las relaciones evolutivas entre proteínas no pueden ser fácilmente detectadas por técnicas estándares de alineamiento de secuencias. El alineamiento estructural puede usarse, por lo tanto, para sugerir relaciones evolutivas entre proteínas que comparten una secuencia común muy corta. Sin embargo, el uso de los resultados como evidencia de un ancestro evolutivo común debe realizarse con cautela dados los posibles efectos de confusión con la evolución convergente, según la cual múltiples secuencias de aminoácidos sin relación filogenética entre sí convergen a una misma estructura terciaria.
Los alineamientos estructurales pueden comparar dos o múltiples secuencias. Puesto que estos alineamientos dependen de información sobre todas las conformaciones tridimensionales de las secuencias problema, el método sólo puede ser usado sobre secuencias donde estas estructuras sean conocidas. Estas se encuentran normalmente por cristalografía de rayos X o espectroscopia de resonancia magnética nuclear. Es posible realizar un alineamiento estructural sobre estructuras producidas mediante métodos de predicción de estructura. En efecto, la evaluación de tales predicciones requiere a menudo un alineamiento estructural entre el modelo y la estructura real conocida para evaluar la calidad del modelo. Los alineamientos estructurales son especialmente útiles para analizar datos surgidos de los campos de la genómica estructural y de la proteómica, y pueden usarse como puntos de comparación para evaluar alineamientos generados por métodos bioinformáticos basados exclusivamente en secuencias.[1]
El resultado de un alineamiento estructural es una superposición de los conjuntos de coordenadas atómicas, así como una distancia media cuadrática mínima (o RMSD, de Root Mean Square Deviation, o desviación de la media cuadrática) entre las estructuras básicas de las proteínas superpuestas. La RMSD de estructuras alineadas indica las divergencias entre ellas. El alineamiento estructural puede complicarse por la existencia de múltiples dominios proteicos en el interior de una o más de las estructuras de entrada, ya que cambios en la orientación relativa de los dominios entre dos estructuras a alinear pueden exagerar la RMSD artificialmente.
Contenido
Datos producidos por el alineamiento estructural
La información mínima producida por un alineamiento estructural correcto es un conjunto de coordenadas tridimensionales superpuestas para cada estructura inicial. Nótese que uno de los elementos de entrada puede estar fijado como referencia y que, por lo tanto, sus coordenadas superpuestas no cambiarán. Las estructuras encajadas pueden usarse para calcular valores RMSD mutuos, así como otras medidas de similitud estructural más sofisticadas como el test de distancia global (GDT,[2] de sus siglas en inglés, y que es la métrica utilizada en CASP, Critical Assessment of Techniques for Protein Structure Prediction). Un alineamiento estructural también implica un alineamiento de secuencias unidimensional desde el que una secuencia identidad, o el porcentaje de residuos que son idénticos entre las estructuras de entrada, puede calcularse como una medida de cuán cercanamente se encuentran ambas secuencias.
Tipos de comparaciones
Puesto que las estructuras de las proteínas se componen de aminoácidos cuyas cadenas laterales están enlazadas por un esqueleto de proteínas comunes, se puede utilizar un número de los posibles subconjuntos diferentes de átomos que conforman una macromolécula de proteína para producir un alineamiento estructural y calcular los correspondientes valores RMSD. Cuando se alinean estructuras con secuencias muy diferentes, los átomos de la cadena lateral, generalmente, no se toman en cuenta, ya que sus identidades difieren en muchos de los residuos alineados. Por esta razón, en los métodos de alineamiento estructural es común usar por defecto sólo los átomos del esqueleto incluidos en el enlace peptídico. Por simplicidad y eficiencia a menudo sólo se consideran las posiciones del carbono alfa, ya que el enlace peptídico tiene una conformación planar mínimamente variante. Sólo cuando las estructuras a alinear son altamente similares, e incluso idénticas, es significativo alinear posiciones de átomos de la cadena lateral, en cuyo caso la RMSD refleja no sólo la conformación del esqueleto de la proteína, sino también los estados de las rotaciones angulares en las cadenas laterales. Otros criterios de comparación que reducen el ruido e impulsan las coincidencias incluyen tomar en consideración la estructura secundaria de las proteínas, los mapas de contactos nativos o patrones de interacción entre residuos, medidas del empaquetamiento de la cadena lateral, y medidas del mantenimiento de los enlaces de hidrógeno.[3]
Superposición estructural
La comparación más sencilla posible entre estructuras de proteínas no intenta alinear las estructuras de entrada, sino que necesita un alineamiento precalculado como input para determinar cuáles de los residuos en la secuencia deben considerarse para el cálculo de la RMSD. La superposición estructural se usa comúnmente para comparar conformaciones múltiples de la misma proteína (en cuyo caso no es necesario el alineamiento ya que la secuencia es la misma) y para evaluar la calidad de los alineamientos producidos usando sólo información de las secuencias entre dos o más secuencias cuyas estructuras son conocidas. Este método utiliza tradicionalmente un sencillo algoritmo de ajuste por mínimos cuadrados, en el que las rotaciones y translaciones óptimas se encuentran minimizando la suma de los cuadrados de las distancias entre todas las estructuras de la superposición.[4] Más recientemente, los métodos bayesianos y de máxima verosimilitud han incrementado enormemente la precisión de las rotaciones, translaciones y matrices de covarianza estimadas para la superposición.[5] [6]
Se han desarrollado algoritmos basados en rotaciones multidimensionales y cuaterniones modificados para identificar relaciones topológicas entre estructuras proteicas sin la necesidad de un alineamiento predeterminado. Tales algoritmos han identificado exitosamente plegamientos canónicos tales como el haz de cuatro hélices alfa.[7] El método SuperPose es suficientemente extensible de ajustar para rotaciones de dominios relativos y otros problemas estructurales.[8]
Complejidad algorítmica
Tanto el enhebrado óptimo de una secuencia de una proteína sobre una estructura conocida[9] como la generación de un alineamiento múltiple de secuencias óptimo[10] han demostrado ser problemas NP-completos. Sin embargo, esto no significa que el problema del alineamiento estructural sea NP-completo. Sobre la base del argumento de que una solución óptima verdadera no es biológicamente significativa debido al error experimental inherente en la determinación de la estructura proteica, se ha desarrollado un algoritmo con aproximadamente tiempo polinómico para el alineamiento de estructuras que produce una familia de soluciones "óptimas" dentro de un parámetro de aproximación para una función de puntuación dada.[11] Sin embargo, con un coste computacional
 para una proteína globular de n residuos, el algoritmo es todavía demasiado costoso computacionalmente para un uso práctico. Como consecuencia, no existen algoritmos prácticos que converjan a las soluciones globales del alineamiento dada una función de puntuación. La mayoría de los algoritmos son, por lo tanto, heurísticos, pero se han desarrollado algunos que garantizan la convergencia a (al menos) maximizadores locales de las funciones de puntuación, y que son prácticos.[12]
para una proteína globular de n residuos, el algoritmo es todavía demasiado costoso computacionalmente para un uso práctico. Como consecuencia, no existen algoritmos prácticos que converjan a las soluciones globales del alineamiento dada una función de puntuación. La mayoría de los algoritmos son, por lo tanto, heurísticos, pero se han desarrollado algunos que garantizan la convergencia a (al menos) maximizadores locales de las funciones de puntuación, y que son prácticos.[12]Representación de estructuras
Las estructuras de las proteínas deben ser representadas en algún espacio de coordenadas independientes para hacerlas comparables. Normalmente se consigue construyendo una matriz, o serie de matrices, secuencia contra secuencia, y que abarquen métricas comparativas en lugar de distancias absolutas relativas a un espacio de coordenadas fijas. Una representación intuitiva es la matriz de distancias, que es una matriz bidimensional que contiene las distancias de todos los emparejamientos entre algún subconjunto de los átomos de cada estructura (por ejemplo, el carbono alfa). La matriz se incrementa dimensionalmente según se incrementa el número de estructuras a alinear simultáneamente. Reduciendo la proteína a una métrica más tosca, tal como elementos de su estructura secundaria o fragmentos estructurales, pueden también producirse alineamientos prácticos, a pesar de la pérdida de información ocasionada por las distancias descartadas puesto que también se descarta ruido.[13] La elección de una representación que facilite la computación es crítica para el desarrollo de mecanismos de alineamiento eficientes.
Métodos
Las técnicas de alineamiento estructural se han usado en la comparación de estructuras individuales o conjuntos de estructuras, y en la producción de bases de datos de comparaciones "todos contra todos" que miden la divergencia entre cada par de estructuras presente en el Protein Data Bank (PDB). Tales bases de datos se usan para clasificar proteínas de acuerdo a su plegamiento.
 Ilustración de los vectores átomo-a-átomo calculados con SSAP. A partir de estos vectores se construirían una serie de vectores diferencia (por ejemplo, entre (FA) en la proteína 1 y (SI) en la proteína 2). Las dos secuencias se representan sobre las dos dimensiones de una matriz para formar una matriz de diferencias entre las dos proteínas. Se aplica programación dinámica sobre todas las posibles matrices de diferencias para construir una serie de trayectorias óptimas de alineamientos locales, que son sumadas para formar una matriz resumen sobre la cual se ejecuta una segunda ronda de programación dinámica.
Ilustración de los vectores átomo-a-átomo calculados con SSAP. A partir de estos vectores se construirían una serie de vectores diferencia (por ejemplo, entre (FA) en la proteína 1 y (SI) en la proteína 2). Las dos secuencias se representan sobre las dos dimensiones de una matriz para formar una matriz de diferencias entre las dos proteínas. Se aplica programación dinámica sobre todas las posibles matrices de diferencias para construir una serie de trayectorias óptimas de alineamientos locales, que son sumadas para formar una matriz resumen sobre la cual se ejecuta una segunda ronda de programación dinámica.DALI
Un método de alineamiento estructural común y popular es DALI (de Distance ALIgnment matrix, o matriz de alineamiento de distancias), que rompe las estructuras problema en fragmentos de hexapéptidos y calcula una matriz de distancia evaluando los patrones de contacto entre fragmentos sucesivos.[14] Las peculiaridades de la estructura secundaria que implican residuos contiguos en la secuencia aparecen en la diagonal principal de la matriz; otras diagonales en la matriz reflejan contactos espaciales entre residuos que no están cercanos uno al otro en la secuencia. Cuando estas diagonales son paralelas a la diagonal principal, las características que representan son paralelas; cuando son perpendiculares, sus características son antiparalelas. Esta representación es intensiva en memoria, ya que las características en la matriz cuadrada son simétricas sobre la diagonal principal (y por lo tanto redundantes).
Cuando las matrices de distancia de dos proteínas comparten las mismas o similares características en aproximadamente las mismas posiciones, puede decirse que tienen similares plegamientos con bucles de longitud similar conectando sus elementos de estructura secundaria. El proceso de alineamiento real de DALI requiere una búsqueda de similitud después de que las matrices de distancia de las dos proteínas se hayan construido; esto se dirige normalmente vía una serie de submatrices superpuestas de tamaño 6x6. Las coincidencias en las submatrices se reúnen en un alineamiento final realizado con un algoritmo estándar de maximización de puntuación. La versión original de DALI usaba una simulación por el método de Montecarlo para maximizar una puntuación de similitud estructural que es función de las distancias entre átomos supuestamente correspondientes. En particular, los átomos más distantes dentro de los elementos característicos vinculados son infraponderados para reducir los efectos del ruido introducido por la movilidad de los bucles, las torsiones de las hélices, y otras variaciones estructurales menores.[13] Dado que DALI se basa en una matriz de distancias "todos contra todos", puede considerar la posibilidad de que las características alineadas estructuralmente podrían aparecer en órdenes diferentes dentro de las dos secuencias en comparación.
El método DALI ha sido también usado para construir una base de datos conocida como FSSP (Fold classification based on Structure-Structure alignment of Proteins, o clasificación de plegamientos basada en alineamientos estructurales de proteínas, y también de Families of Structurally Similar Proteins, o familias de proteínas estructuralmente similares), en la que todas las estructuras de proteínas conocidas son alineadas unas con otras para determinar sus vecinas estructurales y la clasificación de los plegamientos. Hay una base de datos utilizable basada en DALI y mantenida por el EBI, así como un programa descargable y un buscador web basados en una versión autónoma conocida como DaliLite.
SSAP
El método SSAP (de Sequential Structure Alignment Program, o programa de alineamiento secuencial de estructuras) usa doble programación dinámica para generar un alineamiento estructural basado en vectores átomo-a-átomo en un espacio de estructura. En lugar de los carbonos alfa utilizados normalmente en alineamiento estructural, SSAP construye sus vectores desde carbonos beta para todos los residuos excepto glicina, método que así toma en consideración el estado rotamérico de cada residuo así como su localización a lo largo del esqueleto. SSAP trabaja construyendo primero una serie de vectores de distancia inter-residuos entre cada residuo y sus vecinos cercanos no contiguos en cada proteína. Se construyen entonces una serie de matrices conteniendo las diferencias de vectores entre vecinos para cada par de residuos para el que se construyeron los vectores. La programación dinámica aplicada a cada matriz resultante determina una serie de alineamientos locales óptimos que son añadidos a una matriz "resumen" a la que se le aplica de nuevo programación dinámica para determinar el alineamiento estructural global.
SSAP producía originalmente sólo alineamientos de parejas, pero desde entonces ha sido extendido también para alineamientos múltiples.[15] Ha sido aplicado de una manera "todos contra todos" para producir un esquema jerárquico de clasificación de plegamientos conocido como CATH (Class, Architecture, Topology, Homology, o clase, arquitectura, topología, homología),[16] que se ha usado para construir la base de datos CATH Protein Structure Classification.
Extensión combinatoria
El método de extensión combinatoria es similar a DALI en que también rompe cada estructura del conjunto problema en una serie de fragmentos que se intentan entonces volver a ensamblar en un alineamiento completo. Una serie de combinaciones de pares de fragmentos, llamados "pares de fragmentos alineados" (o AFPs, por sus siglas en inglés Aligned Fragment Pairs), se usan para definir una matriz de similitud a partir de la cual se genera una trayectoria óptima para identificar el alineamiento final. Como un medio para reducir el espacio de búsqueda necesario y, por lo tanto, incrementar la eficiencia, sólo los AFPs que cumplen un determinado criterio de similitud local se incluyen en la matriz.[17] Son posibles varias métricas de similitud; la definición original del método de extensión combinatoria incluía sólo superposiciones estructurales y distancias inter-residuos, pero se ha expandido desde entonces para incluir propiedades locales del entorno tales como la estructura secundaria, exposición al disolvente, patrones de puentes de hidrógeno, y ángulos diedros.[17]
Una trayectoria de alineamiento se calcula como la trayectoria óptima a través de la matriz de similitud progresando linealmente a través de las secuencias y extendiendo el alineamiento con el posible siguiente par AFP de más alta puntuación. El par AFP inicial que nuclea el alineamiento puede ocurrir en cualquier punto en la matriz de secuencias. Las extensiones prosiguen entonces con el siguiente AFP que cumple el criterio de distancia dado, restringiendo el alineamiento a tamaños pequeños de gap (o hueco). El tamaño de cada AFP y el tamaño máximo de hueco se requieren como parámetros de entrada, pero normalmente se utilizan los valores empíricamente determinados de 8 y 30, respectivamente.[17] Como DALI y SSAP, la extensión combinatoria se ha usado para construir una base de datas de clasificación de plegamientos a partir de las estructuras conocidas de proteínas en el Protein Data Bank.
MAMMOTH
De sus siglas en inglés MAtching Molecular Models Obtained from Theory, modelos moleculares coincidentes obtenidos de la teoría. Los métodos de alineamiento de estructuras basados en MAMMOTH descomponen la estructura de la proteína en péptidos cortos (heptapéptidos), que son comparados con los heptapéptidos de otra proteína. Se calcula una puntuación de similitud entre dos heptapéptidos usando un método de vector unidad RMS (URMS).[18] Estas puntuaciones se almacenan en una matriz de similitud, y con programación dinámica híbrida (local-global), se calcula el alineamiento de residuos óptimo. Las puntuaciones de similitud entre proteínas calculadas con MAMMOTH se derivan de la probabilidad de obtener un alineamiento estructural dado por casualidad.[19] Este método ha sido optimizado para velocidad y precisión, y es adecuado para estudios estructurales genómicos a gran escala. MAMMOTH se usa en Rosetta@home.
MAMMOTH-mult es una extensión del algoritmo MAMMOTH para ser usado al alinear familias relacionadas de estructuras de proteínas. Este algoritmo es muy rápido y produce alineamientos estructurales consistentes y de alta calidad.[20] Los alineamientos estructurales múltiples calculados con MAMMOTH-mult producen alineamientos de secuencias estructuralmente implícitos, que pueden ser usados posteriormente como plantillas para modelado por homología, predicción de estructura de proteínas por modelos ocultos de Márkov, y búsquedas por tipo de perfil con PSI-BLAST.
RAPIDO
Del inglés Rapid Alignment of Proteins In terms of DOmains (o alineamiento rápido de proteínas en términos de dominios). RAPIDO[21] es un servidor web para el alineamiento 3D de estructuras cristalinas de diferentes moléculas de proteínas, en presencia de cambios conformacionales. Similar a lo que se hace como primer paso en la extensión combinatoria, RAPIDO identifica fragmentos que son estructuralmente similares en las dos proteínas usando una aproximación basada en matrices de distancia por diferencias. Los pares de fragmentos coincidentes, o MFPs (de Matching Fragment Pairs), se representan entonces como nodos en un grafo, los cuales se encadenan juntos para formar un alineamiento por medio de un algoritmo para la identificación de la trayectoria más larga en un grafo acíclico dirigido. El paso final de refinamiento se realiza para mejorar la calidad del alineamiento. Tras alinear las dos estructuras, el servidor aplica un algoritmo genético para la identificación de regiones conformacionalmente invariantes.[22] Estas regiones corresponden a grupos de átomos cuyas distancias interatómicas son constantes (dentro de una tolerancia definida). Haciéndolo así, RAPIDO toma en consideración la variación en la fiabilidad de las coordenadas atómicas empleando funciones de ponderación basadas en los B-valores refinados. Las regiones identificadas por RAPIDO como conformacionalmente invariantes representan conjuntos confiables de átomos para la superposición de las dos estructuras, y que pueden usarse para un detallado análisis de cambios en la conformación. En adición a las funcionalidades proporcionadas por las actuales herramientas, RAPIDO puede identificar regiones estructuralmente equivalentes aun cuando éstas consistan en fragmentos que estén distantes en términos de secuencia y separados por otros dominios móviles.
Desarrollos recientes
Mejorar los métodos de alineamiento estructural constituye un área activa de investigación, y a menudo se proponen métodos nuevos o modificados que pregonan ofrecer ventajas sobre las anteriores y más ampliamente distribuidas técnicas. Un ejemplo reciente, TM-align, utiliza un novedoso método para ponderar su matriz de distancias, en el cual se aplica programación dinámica.[23] [24] La ponderación se propone para acelerar la convergencia (resultado) de la programación dinámica, así como para corregir los efectos vinculados a las longitudes del alineamiento. En un estudio comparativo, TM-align ha resultado mejor, tanto en velocidad como en precisión, que DALI o la extensión combinatoria.[23]
Alineamiento estructural de ARN
Las técnicas de alineamiento estructural se han aplicado tradicionalmente de forma exclusiva a las proteínas, como macromoléculas biológicas fundamentales que asumen estructuras tridimensionales características. Sin embargo, las largas moléculas de ARN también forman estructuras terciarias características que son intermediadas fundamentalmente por puentes de hidrógeno formados entre pares de bases, además de por el apilamiento de las mismas. Las moléculas de ARN no codificante funcionalmente similares pueden resultar especialmente difíciles de extraer de los datos genómicos porque la estructura está más fuertemente conservada que la secuencia tanto en el ARN como en las proteínas,[25] y el más limitado alfabeto del ARN disminuye el contenido de información de cualquier nucleótido dado en cualquier posición particular.
Un método reciente para alineamiento estructural de pares de secuencias de ARN con baja identidad secuencial ha sido publicado e implementado en el programa FOLDALIGN.[26] Sin embargo, este método no es verdaderamente análogo a las técnicas de alineamiento estructural de proteínas, ya que predice computacionalmente las estructuras de las secuencias de ARN de entrada en lugar de requerir estructuras experimentalmente determinadas como entrada. A pesar de que la predicción computacional del proceso de plegamiento de proteínas no ha sido particularmente exitosa hasta la fecha, las estructuras de ARN sin seudonudos pueden ser acertadamente predichas a menudo utilizando métodos de puntuación basados en la energía libre termodinámica que toman en cuenta el emparejamiento y apilamiento de bases.[27]
Software
Elegir una herramienta software para alineamiento estructural puede constituir un desafío debido a la gran variedad de paquetes disponibles, que se diferencian significativamente en metodología y fiabilidad. Debido a su integración con otras herramientas basadas en web del European Bioinformatics Institute, el servidor DaliLite EBI DALI tiene ventaja en la producción de alineamientos estructurales sencillos para investigadores interesados en usar los alineamientos como guía para el trabajo experimental (en lugar de estudiar los métodos de alineamiento por sí mismos). Otro método EBI útil es el Secondary Structure Matcher, que requiere la presencia de al menos dos elementos de estructura secundaria. Una lista más completa de software de alineamiento estructural disponible actualmente y libremente distribuido se puede encontrar en el anexo Software para alineamiento estructural.
Referencias
- ↑ Zhang Y, Skolnick J. (2005). The protein structure prediction problem could be solved using the current PDB library. Proc Natl Acad Sci USA 102(4):1029-34.
- ↑ Zemla A. (2003). LGA - A Method for Finding 3-D Similarities in Protein Structures. Nucleic Acids Research, 31(13):3370-3374.
- ↑ Godzik A. (1996). The structural alignment of proteins: is there a unique answer? Protein Sci 5:1325-8.
- ↑ Martin ACR. http://www.bioinf.org.uk/software/profit/doc/ The software package ProFit (implements McLachlan, AD (1982). Rapid Comparison of Protein Structures. Acta Cryst A38, 871-873.)
- ↑ Theobald DL, Wuttke DS (2006). Empirical Bayes hierarchical models for regularizing maximum likelihood estimation in the matrix Gaussian Procrustes problem. Proceedings of the National Academy of Sciences 103(49):18521-18527. Open Access
- ↑ Theobald DL, Wuttke DS (2006). THESEUS: Maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 22(17):2171-2172. Open Access http://www.theseus3d.org/
- ↑ Diederichs K. (1995). Structural superposition of proteins with unknown alignment and detection of topological similarity using a six-dimensional search algorithm. Proteins 23(2):187-95.
- ↑ Maiti R, Van Domselaar GH, Zhang H, Wishart DS. (2004). SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res 32(Web Server issue):W590-4.
- ↑ Lathrop RH. (1994). The protein threading problem with sequence amino acid interaction preferences is NP-complete. Protein Eng 7(9):1059-68.
- ↑ Wang L, Jiang T. (1994) On the complexity of multiple sequence alignment. J Comput Biol 1:337-348.
- ↑ Kolodny R, Linial N. (2004). Approximate protein structural alignment in polynomial time PNAS 101(33): 12201-12206.
- ↑ Martinez L, Andreani, R, Martinez, JM. (2007). Convergent algorithms for protein structural alignment BMC Bioinformatics 8:306.
- ↑ a b Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
- ↑ Holm L, Sander C (1996). Mapping the protein universe. Science 273: 595-603.
- ↑ Taylor WR, Flores TP, Orengo CA. (1994). Multiple protein structure alignment. Protein Sci 3(10):1858-70.
- ↑ Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. (1997) CATH: A hierarchical classification of protein domain structures. Structure 5(8): 1093-1108.
- ↑ a b c Shindyalov IN, Bourne PE (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11(9) 739-747.
- ↑ Kedem, K., Chew, L., and Elber, R. 1999. Unit-vector RMS (URMS) as a tool to analyze molecular dynamics trajectories. Proteins 37: 554–564
- ↑ Ortiz, A. R., C. E. Strauss, and O. Olmea (2002, November). Mammoth (matching molecular models obtained from theory): An automated method for model comparison. Protein Sci 11 (11), 2606-2621.
- ↑ Lupyan, D., A. Leo-Macias, and A. R. Ortiz (2005, August). A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics 21 (15), 3255-3263.
- ↑ Mosca, Schneider TR. (2008) RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes, Nucleic Acids Research, doi: 10.1093/nar/gkn197 Open Access RAPIDO server
- ↑ Schneider. (2002) A genetic algorithm for the identification of conformationally invariant regions in protein molecules. Acta Crystallogr D Biol Crystallogr vol. 58 (Pt 2) pp. 195-208
- ↑ a b Zhang Y, Skolnick J. (2005) TM-align: A protein structure alignment algorithm based on TM-score. Nucleic Acids Research 33: 2302-2309.
- ↑ Zhang Y, Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57: 702-710.
- ↑ Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J. (2006). Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J. Genome Res 16(7):885-9.
- ↑ Havgaard JH, Lyngso RB, Stormo GD, Gorodkin J. (2005). Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 21(9):1815-24.
- ↑ Mathews DH, Turner DH. (2006). Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol 16(3):270-8.
Lecturas adicionales
- Bourne PE, Shindyalov IN. (2003): Structure Comparison and Alignment. In: Bourne, P.E., Weissig, H. (Eds): Structural Bioinformatics. Hoboken NJ: Wiley-Liss. ISBN 0-471-20200-2
- Yuan X, Bystroff C.(2004) "Non-sequential Structure-based Alignments Reveal Topology-independent Core Packing Arrangements in Proteins", Bioinformatics. Nov 5, 2004
- Jung J, Lee B. (2000). Protein structure alignment using environmental profiles. Protein Eng 13:535-543.
- Ye Y, Godzik A. (2005). Multiple flexible structure alignment using partial order graphs Bioinformatics 21(10): 2362-2369 Abstract
- Sippl M, Wiederstein M (2008). A note on difficult structure alignment problems. Bioinformatics 24(3): 426-427 Full Text
Véase también
Wikimedia foundation. 2010.